
Differential Expression of Metabotropic Glutamate Receptors 2 and 3 in Schizophrenia: A Mechanism for Antipsychotic Drug Action?
Abstract
Objective: Preclinical and clinical data implicate the group II metabotropic glutamate receptors mGluR2 and mGluR3 in the pathophysiology of schizophrenia. Moreover, a recent phase II clinical trial demonstrated the antipsychotic efficacy of a mGluR2/mGluR3 agonist. The purpose of the present study was to distinguish the expression of mGluR2 and mGluR3 receptor proteins in schizophrenia and to quantify glutamate carboxypeptidase II (GCP II) in order to explore a role for the metabotropic receptors in schizophrenia therapeutics. GCP II is an enzyme that metabolizes N -acetyl-aspartyl-glutamate (NAAG), which is the only known specific endogenous agonist of mGluR3 in the mammalian brain. Method: The normal expression levels of mGluR2, mGluR3, and GCP II were determined for 10 regions of the postmortem human brain using specific antibodies. Differences in expression levels of each protein were examined in the dorsolateral prefrontal cortex, temporal cortex, and motor cortex in 15 postmortem schizophrenia subjects and 15 postmortem matched normal comparison subjects. Chronic antipsychotic treatment in rodents was conducted to examine the potential effect of antipsychotic drugs on expression of the three proteins. Results: Findings revealed a significant increase in GCP II protein and a reduction in mGluR3 protein in the dorsolateral prefrontal cortex in schizophrenia subjects, with mGluR2 protein levels unchanged. Chronic antipsychotic treatment in rodents did not influence GCP II or mGluR3 levels. Conclusions: Increased GCP II expression and low mGluR3 expression in the dorsolateral prefrontal cortex suggest that NAAG-mediated signaling is impaired in this brain region in schizophrenia. Further, these data implicate the mGluR3 receptor in the antipsychotic action of mGluR2/mGluR3 agonists.
A role for glutamate in the pathophysiology of schizophrenia is suggested by the psychomimetic properties of phencyclidine. Phencyclidine and ketamine, which are antagonists of the glutamate N -methyl- d -aspartate (NMDA) receptor, can cause positive, negative, and cognitive symptoms reminiscent of schizophrenia in healthy comparison subjects and exacerbate symptoms in individuals with schizophrenia (1 , 2) . The metabotropic glutamate receptors mGluR2 (or GRM2) and mGluR3 (or GRM3), which are differentially distributed at the presynaptic, postsynaptic, and glial compartments, have modulatory roles and influence NMDA receptor function. Molecular studies in animal models of schizophrenia (3) and human postmortem brain tissue (4) support the hypothesis that reduced signaling at the postsynaptic NMDA receptor is linked to schizophrenia. The most convincing evidence of mGluR2 and mGluR3 involvement in schizophrenia was demonstrated in a recent clinical trial using LY2140023, a novel mGluR2/mGluR3 agonist that exhibits potent antipsychotic activity in schizophrenia (5) .
Since LY2140023 is an agonist at both mGluR2 and mGluR3, it is not known whether the antipsychotic effects of this drug are mediated via mGluR2, mGluR3, or both receptors. Human genetic, imaging, and behavioral data suggest that mGluR3 plays an important role in schizophrenia (6 , 7) , while studies using mGluR2 and mGluR3 knockout mice implicate mGluR2 (8) . The respective roles of mGluR2 and mGluR3 in schizophrenia remain unclear. The determination of mGluR2 and mGluR3 expression in high-quality human postmortem brain tissue of individuals with schizophrenia could localize alterations in mGluR2 and/or mGluR3 and suggest a site of action for the mGluR2/mGluR3 agonist.
The endogenous neuropeptide N -acetyl-aspartyl-glutamate (NAAG), which is the substrate of glutamate carboxypeptidase II (GCP II), is prevalent and widely distributed in the mammalian brain (9) . It is a selective agonist for mGluR3 and may also be a weak antagonist at the NMDA receptor, depending on cell type and NMDA receptor subunit composition (9) . NAAG is released into the synapse and metabolized by astrocytic GCP II to N -acetyl aspartate and glutamate. GCP II inhibition reverses phencyclidine-induced behaviors in rodents, presumably by increasing NAAG (10) .
Previous studies examining the combined expression of mGluR2/mGluR3 protein expression in schizophrenia reported either no change (11) or an increase in the prefrontal cortex (12) . In the present study, we determined the individual expression of mGluR2, mGluR3, and GCP II in the dorsolateral prefrontal cortex, temporal lobe cortex, and motor cortex from human postmortem brain tissue samples of 15 schizophrenia subjects and 15 matched normal comparison subjects.
Method
Antibodies
The GCP II antibody was provided by Drs. Joseph Neale and Tomasz Bzdega (Georgetown University, Washington, DC). Specific antibodies for mGluR2 and mGluR3 were obtained from Abcam (Cambridge, Mass.). The mGluR2 antibody (cat#ab52176) is an affinity-purified antibody produced against an epitope-specific immunogen, and its specificity has been tested by Western blot analysis and pre-adsorption studies. The mGluR3 antibody was generated using a synthetic human peptide predicted not to cross-react with mGluR2. Western blot, pre-adsorption, and coimmunoprecipitation studies using mGluR2 and mGluR3 chimeras have demonstrated specificity of this antibody for mGluR3 (13) . The antibody against beta tubulin (cat#MAB3408) was obtained from Millipore (Billerica, Mass.), and the horseradish peroxidase-coupled secondary antibodies were obtained from Vector Laboratories (Burlingame, Calif.).
Human Postmortem Tissue
Characteristics of schizophrenia subjects
Human brain tissue was obtained from the Dallas Brain Collection (14) . Tissue was collected after consent from the subjects’ next of kin was received, along with permission to access medical records and to hold a direct telephone interview with a primary caregiver. All clinical information obtained for each subject was reviewed by three research psychiatrists, and diagnoses were made using DSM-IV criteria. Blood toxicology screens for drugs of abuse, alcohol, and prescription drugs, including psychotropics, were conducted for each subject. Subjects with a known history of neurological disorders or axis I psychiatric disorders other than schizophrenia were excluded. The collection of human brain tissue was approved by the University of Texas Southwestern Medical Center Institutional Review Board. The tissue cohort consisted of 15 pairs of schizophrenia subjects and normal comparison subjects matched as closely as possible for age, brain pH, postmortem interval, and RNA integrity number ( Table 1 [also see the data supplement accompanying the online version of this article]). Four additional normal comparison subjects were used to determine the regional distribution of mGluR2 and mGluR3 in the human brain (see the data supplement accompanying the online version of this article).

Tissue preparation
At the time of brain dissection, tissue samples, including the whole cortical gray matter mantle, were taken from the dorsolateral prefrontal cortex (Brodmann’s area 9), anterior pole of the temporal cortex (Brodmann’s area 38), anterior cingulate cortex (Brodmann’s area 24), orbitofrontal cortex (Brodmann’s area 11), parietal cortex (Brodmann’s area 7), occipital cortex (Brodmann’s area 19), and motor cortex (Brodmann’s area 4). Samples were also dissected from the nucleus accumbens, caudate nucleus, posterior thalamus (pulvinar), hippocampus, and cerebellum. Samples were removed, frozen immediately in a mixture of dry ice and isopentane (1:1, v:v), and stored at –80°C until further use.
Western immunoblotting with scanning densitometry
Tissue samples were removed from storage at –80°C, pulverized on dry ice, and homogenized in buffer (1X phosphate-buffered saline containing 1% sodium dodecyl sulfate, 1 mM phenylmethylsulphonyl fluoride, 20 mg/ml leupeptin, 10 mg/ml pepstatin A, 2 mg/ml aprotinin). Twenty μg of protein per sample were loaded in duplicate on a 10% polyacrylamide gel, transferred to nitrocellulose membranes, blocked for 30 minutes at room temperature (5% nonfat dry milk, 0.1% Tween 20, 50 mM Tris-buffered saline; pH 7.5), and then incubated overnight at 4°C with mGluR2 (1:500 dilution), mGluR3 (1:750 dilution), or GCP II (1:20 dilution) antibody. After washing, blots were incubated with a secondary antibody (1:10,000 dilution, goat antirabbit IgG) for 30 minutes. Immunoreactive proteins were detected with enhanced chemiluminescence (Amersham, N.J.) using Fuji film (LightLabs Company, Dusseldorf, Germany). Film-based images of immunoblots were scanned, and immunopositive bands were quantified using ImageQuant software (Amersham, U.K.) blind to diagnosis. For loading control, b tubulin was utilized.
Brain pH and RNA integrity number determination
Determination for brain pH and RNA integrity number was conducted as described previously (14) . Averages and ranges of tissue quality measures are provided for all subjects ( Table 1 [also see the data supplement accompanying the online version of this article]).
Chronic Antipsychotic Treatment in Rodents
To determine potential effects of chronic antipsychotic treatment on GCP II and mGluR3, Sprague-Dawley rats (Charles River, Wilmington, Mass.) were given a first-generation antipsychotic (haloperidol [N=10]), second-generation antipsychotic (risperidone [N=10]), or placebo (N=10) via drinking water continuously for 6 months as previously described (15) . The rats were then sacrificed, trunk blood collected, and plasma frozen at –20°C until analysis. Drug levels were measured in the laboratory of Dr. Thomas Cooper as previously described (15) . The rodent brains were rapidly removed, and the frontal cortex was dissected and frozen by immersion in –40°C isopentane.
Statistical Analysis
The demographic variables of brain pH, postmortem interval, RNA integrity number, and age were compared between the two groups using t tests. Correlations between protein levels and age, RNA integrity number, and postmortem interval were run with a Spearman rank order correlation. The effect of diagnosis on each protein species was analyzed using a mixed-model analysis of variance (ANOVA), with diagnosis as the between-group factor and brain area (dorsolateral prefrontal cortex, temporal cortex, motor cortex) as the within-group factor. Significant findings were further analyzed using post hoc t tests. When a correlation was found between a demographic variable and protein levels, analysis of covariance (ANCOVA) was performed to correct for the confounding variable. All statistical tests were two-tailed, and p values <0.05 were considered statistically significant. Values outside of two standard deviations from the mean were considered outliers and therefore not included in the statistical analyses.
Results
Regional Distribution of mGluR2 and mGluR3 Protein Expressions in Humans
Four normal subjects (age: 32.5 [SD=9.7] years, postmortem interval: 19.5 [SD=5.3] hours, RNA integrity number: 8.35 [SD=0.61]) were used to determine the regional distribution of mGluR2 and mGluR3 in the brain ( Figure 1 ). High levels of mGluR2 expression were observed in the dorsolateral prefrontal cortex, anterior cingulate cortex, orbitofrontal cortex, parietal cortex, and occipital cortex. Lower levels of expression were detected in the caudate nucleus, nucleus accumbens, and cerebellum, with the lowest levels in the thalamus. The hippocampus expressed relatively high levels of mGluR2. The expression of mGluR3, in general, was more evenly distributed within cortical regions, and the hippocampus expressed the highest levels of mGluR3. In contrast to mGluR2, high levels of mGluR3 were seen in the caudate nucleus and nucleus accumbens, with moderate levels in the thalamus and cerebellum. As a result of the relatively high expression of both mGluR2 and mGluR3 in the cortex, we selected two cortical regions consistently implicated in schizophrenia (dorsolateral prefrontal cortex and temporal cortex) to determine differences in protein expression in the illness. The motor cortex was added as a putative control region.

a The graph illustrates levels of mGluR2 and mGluR3 in 10 brain regions. PFC=dorsolateral prefrontal cortex; ACC=anterior cingulate cortex; OFC=orbitofrontal cortex; PC=parietal cortex; CN=caudate nucleus; NAc=nucleus accumbens; TH=thalamus; HC=hippocampus; CB=cerebellum; OCC=occipital cortex. Bars represent the standard deviations of the mean values.
Comparison Between Schizophrenia Subjects and Normal Comparison Subjects
Fifteen schizophrenia subjects and 15 normal comparison subjects were used in the analysis of GCP II, mGluR2, and mGluR3 protein expression in the dorsolateral prefrontal cortex, temporal cortex, and motor cortex. There were no significant differences between diagnostic groups with respect to age, postmortem interval, brain pH, or RNA integrity number. No significant correlations were found between mGluR2 or mGluR3 and any demographic variable (all r values between –0.13 and 0.33, all p values >0.18) except between mGluR2 and RNA integrity number in the dorsolateral prefrontal cortex (r=0.56, p=0.01). GCP II levels showed a significant correlation with postmortem interval in the dorsolateral prefrontal cortex (r=0.37, p=0.04) and brain pH (r=0.61, p<0.001) in the temporal cortex.
Glutamate carboxypeptidase II
Although there was no main effect of diagnosis on GCP II (F=0.08, df=1, 27, p=0.77), an effect of region (F=17.65, df=1, 54, p<0.0001) and an interaction between diagnosis and region (F=4.11, df=1, 52, p=0.01) were found. Post hoc analysis showed that GCP II protein was significantly higher in the dorsolateral prefrontal cortex in schizophrenia subjects (t=2.27, df=1, 26, p=0.03 [ Figure 2 ]). There were two pairs of schizophrenia subjects (C9/S9 and C11/S11) with low RNA integrity number levels. The significance of the results did not change when we conducted analyses without these two pairs of schizophrenia subjects (F=3.2, df=1, 46, p=0.04).

a The graphs illustrate glutamate carboxypeptidase II (GCP II), mGluR3, and mGluR2 protein levels relative to β tubulin (loading control) in the dorsolateral prefrontal cortex (PFC), temporal cortex (TC), and motor cortex (MC) in schizophrenia subjects and normal comparison subjects. Bars represent the standard deviations of the mean values.
*p<0.05.
mGluR3
Although there was no main effect of diagnosis on mGluR3 (F=3.2, df=1, 21, p=0.08), an effect of region (F=186.1, df=1, 42, p<0.0001) and an interaction between diagnosis and region (F=4.82, df=1, 52, p=0.013) were found. Post hoc tests showed that mGluR3 protein was significantly lower in the prefrontal cortex in schizophrenia subjects (t=2.49, df=1, 22, p=0.02 [ Figure 2 ]). Analysis conducted without the two pairs of schizophrenia subjects with low RNA integrity number (C9/S9 and C11/S11) did not change the significance of the results (t=2.55, df=1, 19, p=0.02).
mGluR2
There was no main effect of diagnosis on mGluR2 (F=1.27, df=1, 27, p=0.3), but an effect of region was observed (F=47.1, df=1, 54, p<0.0001). No significant interaction was seen between diagnosis and region (F=1.91, df=1, 54, p=0.16). Since correlations were seen between mGluR2 protein levels and RNA integrity number, an ANOVA covarying for RNA integrity number was performed. The interaction between diagnosis and region remained nonsignificant (F=1.55, df=3, 23, p=0.2). Exploratory analyses showed no significant differences in mGluR2 expression between groups in any of the three regions (see the data supplement accompanying the online version of this article).
Effects of Chronic Antipsychotic Treatment on GCP II and mGluR3 Expression
The question of whether differences in protein expression could be associated with chronic antipsychotic drug treatment was addressed in two different ways. In the first approach, rats were given two different antipsychotics (haloperidol [N=10] and risperidone [N=10]) continuously for 6 months, using doses that produce plasma levels in the human therapeutic range. Tissue from the frontal cortex was taken from the rats that had been receiving an antipsychotic and analyzed for GCP II and mGluR3 protein expression. Haloperidol- and risperidone plus risperidone-9OH levels in plasma from trunk blood collected at the time of decapitation were 6.8 (SD=1.1) ng/ml and 6.2 (SD=3.2) ng/ml, respectively, demonstrating adequate dosing in these animals. There was no effect of antipsychotic treatment on GCP II (F=1.36, df=1, 27, p=0.3) or mGluR3 protein levels (F=1.48, df=1, 25, p=0.20) in the frontal cortex relative to comparison tissue ( Figure 3 ). In addition, differences in protein levels among schizophrenia subjects who were receiving antipsychotic medication at the time of death (N=10) were contrasted with that of schizophrenia subjects who were not receiving antipsychotic medication at the time of death (N=5). Although the number of schizophrenia subjects who had not been receiving an antipsychotic was low, the results support the lack of an antipsychotic effect on these proteins in the prefrontal cortex (GCP II: t=1.62, df=1, 13, p=0.10; mGluR3: t=0.66, df=1, 10, p=0.50 [see the data supplement accompanying the online version of this article]).
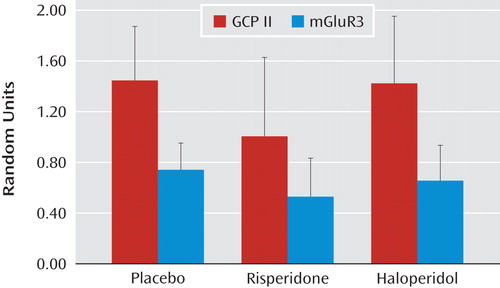
a Graphs illustrate the absence of an effect of chronic treatment in rodents given haloperidol (1.5 mg/kg/day [N=10]) or risperidone (6 mg/kg/day [N=10]) compared with water (placebo) on glutamate carboxypeptidase II (GCP II) and mGluR3 protein expression in the frontal cortex. Bars represent the standard deviations of the mean values.
Discussion
The individual expression profiles of mGluR2 and mGluR3 protein in the human brain have not been previously demonstrated because selective antibodies were lacking. The recent availability of antibodies specific for each of these receptors has made it possible to determine their individual expression in the normal human brain and disease populations. In the present study, we examined the expression profile of these proteins across a range of regions in the normal human brain. We then compared postmortem tissue samples from schizophrenia subjects and matched normal comparison subjects in two brain regions important to the pathophysiology of schizophrenia (dorsolateral prefrontal cortex and temporal cortex) as well as in a putative control region (motor cortex). These analyses revealed that mGluR3 and GCP II are altered in the dorsolateral prefrontal cortex but not the temporal and motor cortices in schizophrenia.
The metabotropic glutamate receptors mGluR2 and mGluR3 have distinct expression patterns in the human brain. Cortical expression of these proteins in humans is similar to that seen in rodents (16 , 17) . In contrast, the human nucleus accumbens and caudate nucleus express relatively low levels of mGluR2 protein and moderate levels of mGluR3 protein compared with high levels of these proteins found in rodents. The lower expression of mGluR2 and mGluR3 in the nucleus accumbens and caudate nucleus—regions that receive projections from dopamine neurons—may be important in light of the modulatory roles of these receptors on dopamine neurotransmission. We previously reported that mGluR2 and mGluR3 mRNAs are expressed in dopamine neurons (18) .
Differences in Schizophrenia
There are several prominent findings in the present study. First, the changes in mGluR3 and GCP II protein expression in schizophrenia were localized to the dorsolateral prefrontal cortex (Brodmann’s area 9), a brain region repeatedly implicated in this illness (19 , 20) . Many studies have shown that volunteers with schizophrenia perform poorly on working memory tasks and show deficits in dorsolateral prefrontal cortex functional activation (19 , 20) . Moreover, molecular studies have identified several replicated alterations in the dorsolateral prefrontal cortex (21) . Our findings are consistent with the localization of molecular alterations to this brain region and extend previous knowledge to the mGluR system. Second, we found a reduction in mGluR3, not mGluR2, leading us to suggest that mGluR3 could be the specific molecular target of the novel mGluR2/mGluR3 agonist LY2140023, mediating its antipsychotic action. Further, both mGluR3 and GCP II are significantly changed in the same brain region, suggesting a potential association between these two proteins, which are already known to be functionally related. NAAG, an endogenous agonist of mGluR3, is the substrate of GCP II. Levels of NAAG are largely determined by GCP II activity, such that GCP II activity may be considered an indirect marker of NAAG levels. Increased GCP II levels indicate low NAAG concentrations (9) . As a result, we reason that NAAG levels are decreased in the dorsolateral prefrontal cortex in schizophrenia. These changes are consistent with phencyclidine studies showing that GCP II inhibition reduces phencyclidine-induced behaviors in laboratory animals (22) . Third, we have provided evidence in the present study that the changes in GCP II and mGluR3 expression are not likely to be secondary to antipsychotic treatment.
Previous Studies of GCP II and mGluR3
GCP II levels and activity in the prefrontal cortex of schizophrenia subjects have been previously examined but with inconsistent results. Some of these studies have reported no change in mRNA expression (23) . Some have reported a decrease in quantitative binding (24) , while others have found reduced enzyme activity (25) . Although these studies examined GCP II in Brodmann’s area 46, in the present study, we used tissue from Brodmann’s area 9. In addition, we report increases in protein expression not in mRNA levels or enzyme activity. Of note, we found an influence of postmortem interval on GCP II levels, which is a variable that may influence results across laboratories. Further, disease heterogeneity in schizophrenia must always be considered.
Expression profiles of mGluR2 and mGluR3 in the dorsolateral prefrontal cortex of schizophrenia subjects have also been previously examined (11 , 12 , 18 , 26 , 27) but with inconsistent results. To the best of our knowledge, at the mRNA level, no changes in mGluR3 in the dorsolateral prefrontal cortex have been reported (18 , 26) , although alterations in expression of a splice variant may exist (27) . Crook et al. (11) did not find any change in the combined expression of mGluR2/mGluR3 protein in Brodmann’s area 46. However, a later study reported an increase in mGluR2/mGluR3 immunoreactivity in Brodmann’s area 46 but not in Brodmann’s area 9. Corti et al. (28) developed a mGluR3-specific antibody and tested it on Brodmann’s area 10 schizophrenia tissue but found no change in the total or monomeric form of mGluR3. However, they did find a decrease in the dimeric form of mGluR3. In the present study, we found a reduced expression of monomeric mGluR3 protein. The apparent discrepancy between the Corti et al. report and our data is not clear. Differences in brain regions analyzed (Brodmann’s area 9 versus Brodmann’s area 10), differences in tissue quality characteristics (such as age and postmortem interval), or differences in assays between laboratories may be contributory factors.
The lack of effect of diagnosis on mGluR2 expression reflects normal levels of mGluR2 protein and is consistent with our previous study, which showed unchanged levels of mGluR2 mRNA in the gray matter of the dorsolateral prefrontal cortex in schizophrenia (18) , but differs from a recent report of a small cohort of seven pairs of medication-free schizophrenia subjects showing reduced mGluR2 mRNA (13) . In the present study, no differences exist between schizophrenia subjects who received drug treatment and those who did not receive drug treatment. However, as a result of the small sample size, further examination may be warranted. We did not find an effect of antipsychotic treatment on mGluR3 or GCP II protein expression, an outcome consistent with previous studies (27 – 29) .
Functional Role of mGluR3 Receptors
The mGluR3 receptors are located at the presynaptic terminal, postsynaptic terminal, and glial cells (30) and have distinct functions at each site. Overall, activation of mGluR3 receptors has been linked to the modulation of a variety of physiological functions, including release of neurotransmitters, regulation of synaptic plasticity, and enhancing neuroprotection (9) . Several mGluR3-mediated effects may be of particular significance in schizophrenia.
Neuronal mGluR3 receptors modulate the glutamate and gamma-aminobutyric acid (GABA) systems, both of which are implicated in the pathophysiology of schizophrenia (9) . Presynaptic mGluR3 receptors inhibit the release of glutamate and GABA into the synapse, while postsynaptic mGluR3 receptors have been shown to modulate NMDA and GABA type A (GABA A ) receptor function (9) . In the prefrontal cortex, the enhancement of NMDA receptor function by mGluR2/mGluR3 receptors (31) , most likely mGluR3 (10) , has been suggested as the physiological basis for reversal of phencyclidine-induced behaviors (10 , 31) . Activation of mGluR3 receptors also regulates GABA A receptor subunit expression, specifically α 1 , α 5 , α 6 , β 2 , γ 2 , and δ subunits ( 32 [unpublished data available upon request from Ghose S.]). In the schizophrenia prefrontal cortex, alterations in several GABA A subunits, including α 1 , γ 2 , and δ, have been reported (33) . It is plausible that the mGluR3 decreases in the dorsolateral prefrontal cortex reported in the present study are related to the GABA A abnormalities in schizophrenia.
Astrocytic mGluR3 receptor activation generates the formation of transforming growth factor β 1 and transforming growth factor β 2(34) , factors that are neuroprotective. The transforming growth factor β pathway proteins modulate synaptic plasticity (35) and glutamatergic function (36) and enhance dendritic growth and spine formation (37) . Moreover, activation of astrocytic mGluR3 receptors plays a role in modulating brain microcirculation (38) . Baslow et al. (38) suggested that NAAG activates astrocytic mGluR3 and triggers Ca 2+ currents that spread to astrocytic end-feet in contact with the vascular system, where a secondary release of vasoactive agents increases blood flow. As a result, we speculate that low NAAG levels are associated with the low levels of regional blood flow often reported in the prefrontal cortex in schizophrenia subjects (39) .
Model of mGluR3 Dysfunction in the Dorsolateral Prefrontal Cortex in Schizophrenia
We propose a formulation of glutamatergic function in the prefrontal cortex of individuals with schizophrenia that recognizes the complex roles of mGluR3 in brain function and is consistent with a putative glutamate reduction in schizophrenia ( Figure 4 ). At the postsynaptic mGluR3 receptor, the reduction in protein would diminish glutamate signal through a loss of positive modulation at the NMDA receptor ( Figure 4 B). Moreover, at the glial mGluR3, reduced receptor activation would decrease transforming growth factor β release. The reduced level of this growth factor may be associated with the smaller size of prefrontal neurons and reduction in dendritic branching reported in schizophrenia. Further, reduced astrocytic mGluR3 activation can lead to the decrease in Ca 2+ signaling and subsequent reduction in local blood flow in the dorsolateral prefrontal cortex of individuals with schizophrenia demonstrated by numerous previous studies in schizophrenia (39) . In addition, the increase in GCP II on glial cells decreases NAAG levels through active degradation, further reducing mGluR3-mediated signaling.

a Panel A shows a schematic representation of the three compartments of the normal synapse (presynaptic terminal, postsynaptic terminal, and glial cell), with localization of mGluR3 and glutamate carboxypeptidase II (GCP II). N -acetyl-aspartyl-glutamate (NAAG) is released from the presynaptic terminal into the synapse, where it can activate mGluR3 receptors located at 1) the postsynaptic terminal to potentiate NMDA responses (+) and regulate GABA A receptor subunit expression, 2) the presynaptic terminal to regulate its own release (–), or 3) the glial cell, where it mediates the release of transforming growth factor β (TGF β) and modulates local microcirculation. Panel B illustrates impaired NAAG-mediated activation at the mGluR3 receptor in schizophrenia. This could reduce NMDA potentiation, alter GABA A receptor composition, release NAAG (which is metabolized by higher GCP II levels), decrease TGF β release, and reduce regional blood flow. Panel C illustrates how mGluR2/3 agonists could reverse the effects of high GCP II and low mGluR3 to restore function at the mGluR3 receptor.
We also propose that activation of mGluR3 by agonists such as LY2140023 would reverse the changes brought about by mGluR3 reduction in the prefrontal cortex of individuals with schizophrenia ( Figure 4 C). Although mGluR3 activation would further reduce NAAG release via presynaptic inhibition, it would generate an increased response at the postsynaptic mGluR3 receptor to potentiate NMDA function. The net effect would be an increase in NMDA-mediated signaling at the glutamate synapse. In addition, disease-associated GABA deficits mediated by low mGluR3 would also be reversed by LY2140023. In the glial compartment, mGluR3 activation would augment transforming growth factor β levels, reversing the potentially deleterious effects on neuronal morphology and function. Further, glial mGluR3 activation could restore local blood flow in the prefrontal cortex.
In summary, LY2140023 could 1) reverse mGluR3-mediated deficits in schizophrenia to increase function at the NMDA receptor, 2) “normalize” GABA function, 3) increase transforming growth factor β, and 4) restore dorsolateral prefrontal cortex perfusion. These interpretations provide a model of prefrontal cortical dysfunction in schizophrenia in which to conceptualize our data and postulate mechanisms for the action of the selective mGluR2/mGluR3 agonist.
Limitations to the Study
The extent to which human postmortem tissue reflects in vivo conditions is always a question but is buttressed in the present study by the selection of high-quality tissue characteristics. Several parameters have been identified to mark tissue quality, such as RNA integrity number and postmortem interval (14) . The tissue used in the present study was of high quality as evaluated by these parameters. Moreover, the potential effects of antemortem antipsychotic treatment on gene expression products can be an important potential confound. Although the present study attempted to address the latter issue using two approaches, and both suggested no chronic medication effect, the possibility of a drug effect must always be considered. Additionally, one cannot exclude the possibility that these drugs have distinct effects in the human brain relative to the rodent brain. We examined protein levels in the gray matter of the cortical regions. The possibility of changes in the dorsolateral prefrontal cortex white matter (18) will need further evaluation. In addition, we are unable to comment on whether the mGluR3 change we found localizes to any particular receptor population (i.e., presynaptic, postsynaptic, or glial), nor can we draw conclusions about the dynamic regulation between GCP II and mGluR3.
In closing, we have provided evidence that NAAG-mediated neurotransmission at the mGluR3 receptor is disrupted in the dorsolateral prefrontal cortex in schizophrenia, based on human postmortem tissue measures of the proteins involved. The defects we have reported could be attenuated by mGluR3 agonists reversing the consequences of the protein changes, putatively ameliorating the symptoms of the illness. This leads us to speculate that this molecular target could mediate the therapeutic response to LY2140023, the first mGluR2/mGluR3 agonist with an antipsychotic action in schizophrenia (5) .
1. Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA: Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 2001; 25:455–467Google Scholar
2. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, Charney DS: Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 1994; 51:199–214Google Scholar
3. Moghaddam B, Adams BW: Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998; 281:1349–1352Google Scholar
4. Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA: Ionotropic glutamate receptors and expression of N-methyl- d -aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry 2000; 157:1141–1149 Google Scholar
5. Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD: Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med 2007; 13:1102–1107Google Scholar
6. Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, Mattay VS, Bertolino A, Hyde TM, Shannon-Weickert C, Akil M, Crook J, Vakkalanka RK, Balkissoon R, Gibbs RA, Kleinman JE, Weinberger DR: Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A 2004; 101:12604–12609Google Scholar
7. Tan HY, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF, Mattay VS, Meyer-Lindenberg A, Weinberger DR, Callicott JH: Epistasis between catechol- o -methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A 2007; 104:12536–12541 Google Scholar
8. Woolley ML, Pemberton DJ, Bate S, Corti C, Jones DN: The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology (Berl) 2008; 196:431–440Google Scholar
9. Neale JH, Bzdega T, Wroblewska B: N-acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J Neurochem 2000; 75:443–452Google Scholar
10. Olszewski RT, Wegorzewska MM, Monteiro AC, Krolikowski KA, Zhou J, Kozikowski AP, Long K, Mastropaolo J, Deutsch SI, Neale JH: Phencyclidine and dizocilpine induced behaviors reduced by N-acetylaspartylglutamate peptidase inhibition via metabotropic glutamate receptors. Biol Psychiatry 2008; 63:86–91Google Scholar
11. Crook JM, Akil M, Law BC, Hyde TM, Kleinman JE: Comparative analysis of group II metabotropic glutamate receptor immunoreactivity in Brodmann’s area 46 of the dorsolateral prefrontal cortex from patients with schizophrenia and normal subjects. Mol Psychiatry 2002; 7:157–164Google Scholar
12. Gupta DS, McCullumsmith RE, Beneyto M, Haroutunian V, Davis KL, Meador-Woodruff JH: Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse 2005; 57:123–131Google Scholar
13. González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, López-Giménez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC: Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008; 452:93–97Google Scholar
14. Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ, Tamminga CA: Human postmortem tissue: What quality markers matter? Brain Res 2006; 1123:1–11Google Scholar
15. Gao XM, Cooper T, Suckow RF, Tamminga CA: Multidose risperidone treatment evaluated in a rodent model of tardive dyskinesia. Neuropsychopharmacology 2006; 31:1864–1868Google Scholar
16. Ohishi H, Neki A, Mizuno N: Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res 1998; 30:65–82Google Scholar
17. Tamaru Y, Nomura S, Mizuno N, Shigemoto R: Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience 2001; 106:481–503Google Scholar
18. Ghose S, Crook JM, Bartus CL, Sherman TG, Herman MM, Hyde TM, Kleinman JE, Akil M: Metabotropic glutamate receptor 2 and 3 gene expression in the human prefrontal cortex and mesencephalon in schizophrenia. Int J Neurosci 2008; 118:1609–1627Google Scholar
19. Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR: Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry 2003; 160:2209–2215Google Scholar
20. Perlstein WM, Carter CS, Noll DC, Cohen JD: Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry 2001; 158:1105–1113Google Scholar
21. Harrison PJ, Weinberger DR: Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 2005; 10:40–68; image 5Google Scholar
22. Olszewski RT, Bukhari N, Zhou J, Kozikowski AP, Wroblewski JT, Shamimi-Noori S, Wroblewska B, Bzdega T, Vicini S, Barton FB, Neale JH: NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J Neurochem 2004; 89:876–885Google Scholar
23. Ghose S, Weickert CS, Colvin SM, Coyle JT, Herman MM, Hyde TM, Kleinman JE: Glutamate carboxypeptidase II gene expression in the human frontal and temporal lobe in schizophrenia. Neuropsychopharmacology 2004; 29:117–125Google Scholar
24. Guilarte TR, Hammoud DA, McGlothan JL, Caffo BS, Foss CA, Kozikowski AP, Pomper MG: Dysregulation of glutamate carboxypeptidase II in psychiatric disease. Schizophr Res 2008; 99:324–332Google Scholar
25. Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE, Coyle JT: Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry 1995; 52:829–836Google Scholar
26. Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC: Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Brain Res Mol Brain Res 1998; 56:207–217Google Scholar
27. Sartorius LJ, Weinberger DR, Hyde TM, Harrison PJ, Kleinman JE, Lipska BK: Expression of a GRM3 splice variant is increased in the dorsolateral prefrontal cortex of individuals carrying a schizophrenia risk SNP. Neuropsychopharmacology 2008; 33:2626–2634Google Scholar
28. Corti C, Crepaldi L, Mion S, Roth AL, Xuereb JH, Ferraguti F: Altered dimerization of metabotropic glutamate receptor 3 in schizophrenia. Biol Psychiatry 2007; 62:747–755Google Scholar
29. Flores C, Coyle JT: Regulation of glutamate carboxypeptidase II function in corticolimbic regions of rat brain by phencyclidine, haloperidol, and clozapine. Neuropsychopharmacology 2003; 28:1227–1234Google Scholar
30. Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ: The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience 1996; 71:949–976Google Scholar
31. Homayoun H, Jackson ME, Moghaddam B: Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J Neurophysiol 2005; 93:1989–2001Google Scholar
32. Ghose S, Wroblewska B, Corsi L, Grayson DR, De Blas AL, Vicini S, Neale JH: N-acetylaspartylglutamate stimulates metabotropic glutamate receptor 3 to regulate expression of the GABA(A) alpha 6 subunit in cerebellar granule cells. J Neurochem 1997; 69:2326–2335 Google Scholar
33. Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA: Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 2008; 13:147–161Google Scholar
34. Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F: Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J Neurosci 1998; 18:9594–9600Google Scholar
35. Fukushima T, Liu RY, Byrne JH: Transforming growth factor-beta 2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007; 17:5–9 Google Scholar
36. Kurisaki A, Inoue I, Kurisaki K, Yamakawa N, Tsuchida K, Sugino H: Activin induces long-lasting N-methyl- d -aspartate receptor activation via scaffolding PDZ protein activin receptor interacting protein 1. Neuroscience 2008; 151:1225–1235 Google Scholar
37. Esquenazi S, Monnerie H, Kaplan P, Le Roux P: BMP-7 and excess glutamate: opposing effects on dendrite growth from cerebral cortical neurons in vitro. Exp Neurol 2002; 176:41–54Google Scholar
38. Baslow MH, Dyakin VV, Nowak KL, Hungund BL, Guilfoyle DN: 2-PMPA, a NAAG peptidase inhibitor, attenuates magnetic resonance BOLD signals in brain of anesthetized mice: evidence of a link between neuron NAAG release and hyperemia. J Mol Neurosci 2005; 26:1–15Google Scholar
39. Hill K, Mann L, Laws KR, Stephenson CM, Nimmo-Smith I, McKenna PJ: Hypofrontality in schizophrenia: a meta-analysis of functional imaging studies. Acta Psychiatr Scand 2004; 110:243–256Google Scholar

