
Neural Substrates of Treatment Response to Cognitive-Behavioral Therapy in Panic Disorder With Agoraphobia
Abstract
Objective
Although exposure-based cognitive-behavioral therapy (CBT) is an effective treatment option for panic disorder with agoraphobia, the neural substrates of treatment response remain unknown. Evidence suggests that panic disorder with agoraphobia is characterized by dysfunctional safety signal processing. Using fear conditioning as a neurofunctional probe, the authors investigated neural baseline characteristics and neuroplastic changes after CBT that were associated with treatment outcome in patients with panic disorder with agoraphobia.
Method
Neural correlates of fear conditioning and extinction were measured using functional MRI before and after a manualized CBT program focusing on behavioral exposure in 49 medication-free patients with a primary diagnosis of panic disorder with agoraphobia. Treatment response was defined as a reduction exceeding 50% in Hamilton Anxiety Rating Scale scores.
Results
At baseline, nonresponders exhibited enhanced activation in the right pregenual anterior cingulate cortex, the hippocampus, and the amygdala in response to a safety signal. While this activation pattern partly resolved in nonresponders after CBT, successful treatment was characterized by increased right hippocampal activation when processing stimulus contingencies. Treatment response was associated with an inhibitory functional coupling between the anterior cingulate cortex and the amygdala that did not change over time.
Conclusions
This study identified brain activation patterns associated with treatment response in patients with panic disorder with agoraphobia. Altered safety signal processing and anterior cingulate cortex-amygdala coupling may indicate individual differences among these patients that determine the effectiveness of exposure-based CBT and associated neuroplastic changes. Findings point to brain networks by which successful CBT in this patient population is mediated.
Although cognitive-behavioral therapy (CBT) is a first-line treatment for panic disorder with agoraphobia (1), little is known about the neural substrates underlying treatment response. Fear conditioning represents a central pathway for the development and maintenance of panic disorder with agoraphobia (2–4). Behavioral studies have found altered safety signal processing (5, 6) and enhanced resistance to extinction (7) in patients with panic disorder. In line with this, neuroimaging studies have indicated altered activation in the right anterior cingulate cortex, the amygdala, and the brainstem in patients with panic disorder during instructed fear conditioning (8), with patients exhibiting increased activation during a safety condition. These findings indicate that a network that signals fear is incorrectly activated in individuals with panic disorder with agoraphobia.
Neuroimaging studies of treatment-related neural changes in patients with panic disorder are rare but provide the first evidence that brain activation is sensitive to change, although results remain highly inconsistent (9–12). It also remains unknown how changes in basic brain metabolism or brain metabolism at rest relate to dysfunctional processes of interest (e.g., fear conditioning). A recent analysis of treatment effects (3), based on the sample in the present study, found neural correlates of enhanced differential conditioning in the left inferior frontal gyrus that attenuated after CBT. Despite this significant reduction, there was increased functional connectivity between the inferior frontal gyrus and limbic structures (e.g., the amygdala and the hippocampus) that remained stable across time. Although these findings contribute to our understanding of the pathophysiology of panic disorder, neural differences among patients determining the effectiveness of CBT and pathways of neuroplastic change underlying successful treatment response have not yet been evaluated.
Given previous evidence for the relevance of altered fear conditioning, safety signal processing, and fronto-limbic connectivity in patients with panic disorder with agoraphobia, we investigated 1) the predictive value brain activation patterns during fear conditioning have for treatment response and 2) changes in brain activation associated with treatment response. We hypothesized that responders and nonresponders would differ 1) in neurofunctional activation in neural networks subserving fear processing (e.g., the amygdala, hippocampus, and anterior cingulate cortex) during safety signal processing, 2) in functional connectivity between fronto-limbic networks, and 3) in pathways of neuroplastic change after CBT.
Method
Participants
The study was part of the National Research Network PANIC-NET encompassing a randomized controlled clinical trial of CBT for panic disorder with agoraphobia (13). Eight German centers participated in the clinical trial (Aachen, Berlin-Adlershof, Berlin-Charité, Bremen, Dresden, Greifswald, Münster, and Würzburg) in which 369 patients who met DSM-IV-TR criteria for panic disorder with agoraphobia received treatment. Four of the centers (Aachen, Berlin-Charité, Dresden, and Münster) also participated in a functional MRI (fMRI) study. Baseline clinical characteristics were comparable between patients in the fMRI study and those in the non-fMRI sample. The manualized treatment protocol consisted of 12 twice-weekly sessions that focused on behavioral exposure in situ. Patients were randomly assigned to one of two CBT arms, which differed only with regard to therapist-guided or nonguided exposure sessions (six out of 12 sessions). Because participants in the treatment arms received comparable treatment and exhibited significant symptom reduction after CBT (13), the groups were collapsed in the present study. The frequency of treatment conditions was similar across responders and nonresponders (see Table S1 in the data supplement that accompanies the online edition of this article). In the clinical trial, treatment response was classified as a reduction >50% from baseline to posttreatment assessment on the primary outcome measure, the score on the Hamilton Anxiety Rating Scale (using the Structured Interview Guide for the Hamilton Anxiety Rating Scale [SIGH-A]) (14). We adopted this criterion to maintain comparability with previous studies (13, 15). The Clinical Global Impressions Scale (CGI) (16), the SIGH-A, the Panic and Agoraphobia Scale (17), the Anxiety Sensitivity Index (18), and the Beck Depression Inventory–II (BDI-II) (19) were used for sample characterization. Inclusion criteria were a primary diagnosis of panic disorder with agoraphobia as assessed by the Computer-Assisted Personal Interview version of the Munich-Composite International Diagnostic Interview (20) and validated by clinical experts, a SIGH-A score ≥18, a CGI score ≥4, and age 18–65 years. Patients were excluded if they were unable to comply with the study schedule; reported clinically significant suicidal intent; met diagnostic criteria for any psychotic or bipolar disorder, borderline personality disorder, or current alcohol dependence; had a medical condition that could explain their symptoms; or fulfilled MRI-related exclusion criteria. Current comorbid diagnoses, including unipolar depression and other anxiety disorders, were allowed unless they were of primary clinical concern. As such, our sample can be considered both to have relatively severe symptoms and to be representative of patients seen in clinical practice. Participants were required to discontinue all psychopharmacological medications, and those who were taking psychotropic medications underwent a 4-week washout period. Participants provided written informed consent after receiving a complete description of the study. The study was approved by all ethics committees of the participating fMRI centers. Of 369 patients enrolled in the clinical trial, 194 were recruited at fMRI centers, and 89 consented to participate in the present study. At baseline, 49 quality-controlled data sets were available; seven patients were excluded at the posttreatment assessment because of nonadherence or insufficient data quality, leaving 42 data sets for analysis of treatment-related changes. A detailed description of measures of quality control in this fMRI multicenter study and the CONSORT diagram for the study have been published elsewhere (3).
Fear-Conditioning Task
Parallel versions of a previously validated (3, 21) differential fear-conditioning task were applied before and after CBT. During differential conditioning, the reinforced conditioned stimulus (CS+) induces fear learning, while the nonreinforced conditioned stimulus, CS–, (which is never followed by the unconditioned stimulus [US]) acquires safety signal properties. The task consisted of three phases: familiarization, with 16 trials of each CS; acquisition, with 32 trials of each CS; and extinction, with 16 trials of each CS. Colored geometric stimuli represented the CSs (presentation time: 2,000 ms, with a variable intertrial interval of 4.785–7.250 seconds). An aversive auditory tone (white noise, 100 ms) was used as the US. It was pseudorandomly paired with one of the CSs during the acquisition phase (counterbalanced among participants). A partial reinforcement rate of 50% was employed. During acquisition, only those trials in which no US was delivered (CS+ unpaired) were analyzed. The task duration was approximately 17 minutes.
fMRI Data Acquisition and Analysis
fMRI images were acquired using 3-T Philips Achieva (Aachen, Münster, Germany), 3-T Siemens Trio (Dresden, Germany), and 3-T General Electric Healthcare (Berlin) scanners. A total of 505 axial functional images (matrix=64×64; 30 slices interleaved; field of view=230; voxel size=3.6×3.6×3.8 mm; TE=30 ms; TR=2 seconds), covering the whole brain and positioned parallel to the intercommissural line (anterior commissure-posterior commissure), were recorded, and a three-dimensional structural data set was used (matrix=128×112; 88 slices; field of view=256; voxel size=2×2×2 mm; TE=3.93 ms; TR=1,100 ms; flip angle=9°). MR images were analyzed with statistical parametric mapping using SPM5 (www.fil.ion.ucl.ac.uk) implemented in MATLAB, version 7.1 (MathWorks, Natick, Mass.). The first five volumes were discarded to minimize T1 saturation effects. A high-pass filter (cutoff period, 128 seconds) was applied to remove low-frequency fluctuations in the blood-oxygen-level-dependent (BOLD) signal. Following slice time correction, functional images were temporally and spatially aligned and normalized into a standard stereotactic space (Montreal Neurological Institute template: 2×2×2 mm). Accounting for differences in intrinsic smoothness between scanners, an iterative smoothness equalization (22) procedure was performed (12-mm full-width-at-half-maximum Gaussian isotropic kernel). Thus, data from all centers were iteratively smoothed until a smoothness of 12 mm full width at half maximum was reached. Assuming an intrinsic smoothness of 4–6 mm, smoothing is comparable to a predefined kernel of 8 mm full width at half maximum in a normal smoothing procedure.
At the single-subject level, realignment parameters were included as regressors to account for movement artifacts. The BOLD response for each event type (CS+ paired, CS+ unpaired, CS–, and US) and phase (familiarization, acquisition, and extinction) was modeled by the canonical hemodynamic response function used in SPM5 within the framework of the general linear model. Parameter estimates (β) and t-statistic images were calculated for each subject. Group analyses were performed by entering contrast images into a flexible factorial analysis, in which subjects were treated as random variables. The first group analysis focused on baseline characteristics and included data for 49 patients (baseline sample). A second analysis of changes from baseline to posttreatment assessment included data sets for 42 patients (baseline and posttreatment assessment sample). Since main effects during the baseline assessment were observed during the extinction phase, treatment-related changes were investigated during extinction. As used in previous analyses (3), the models included an fMRI center variable to account for scanner differences between sites and another covariate on educational level. Responders and nonresponders did not differ in baseline scores on the SIGH-A but did differ in baseline scores on the Panic and Agoraphobia Scale. We aimed to investigate correlates of treatment response independent of differences in panic symptoms and comorbid depressive symptoms, including baseline scores on the Panic and Agoraphobia Scale and BDI-II as covariates. However, results were comparable to those using a model without the Panic and Agoraphobia Scale and BDI-II, revealing that findings were unrelated to panic severity and depressive symptoms at baseline. F contrasts were computed for the interaction effects of group-by-CS (baseline model, separately for the experimental phases), group-by-time, and group-by-CS-by-time (baseline and posttreatment assessment model), followed by post hoc t tests (inclusive of masking by the respective F contrast at a p value <0.005) to explore the direction of effects. As in previous analyses (3), a Monte Carlo simulation of the brain volume was conducted to establish an appropriate voxel contiguity threshold (23). Assuming an individual voxel type I error at a p value <0.005, a cluster extent of 142 contiguous resampled voxels was indicated as sufficient to correct for multiple voxel comparisons at a p value <0.05. For all analyses, voxels with a significance level <0.005, uncorrected, belonging to clusters with at least 142 voxels are reported. Since this correction algorithm could bias findings toward larger brain regions, a region-of-interest analysis of the amygdala was conducted using the Wake Forest University PickAtlas (24) (p<0.05, family-wise-error corrected).
For connectivity analyses, we extracted eigenvectors (adjusted for the effect of movement parameters) from the anterior cingulate cortex cluster on the first level across the entire task, serving as regressors in a separate first-level model. Individual activation maps reflected the correlation of each voxel time course with the time course of the anterior cingulate cortex. These contrast images were used in the group analysis focusing on group differences (49 patients) and group-by-time interactions (42 patients) in connectivity between the anterior cingulate cortex as a seed region and the amygdala (exploratory whole-brain analysis: p<0.005, uncorrected; region-of-interest analysis: p<0.05, family-wise-error corrected).
Classification of treatment response by the magnitude of brain activation at baseline in three clusters of interest (the anterior cingulate cortex, the hippocampus, and the amygdala) and anterior cingluate cortex-amygdala connectivity was tested using a receiver operating characteristic curve analysis. A Bonferroni-corrected alpha set at 0.0125 indicated statistical significance. Analyses were carried out using SPSS, version 19.0 (IBM, Armonk, N.Y.).
Results
Neural Correlates of Treatment Response at Baseline
Demographic and clinical characteristics of the baseline sample are summarized in Table 1. We observed a significant group-by-CS interaction during extinction (Table 2). Post hoc t tests (responders > nonresponders [CS+ unpaired > CS–]) found elevated neural activation encompassing the right pregenual anterior cingulate cortex, the hippocampus, the pre- and postcentral gyri and amygdala (region-of-interest analysis), the left middle temporal gyrus, the fusiform gyrus, and the inferior occipital gyrus. Beta values indicated that this interaction effect was driven by a pronounced increase in activation during CS– processing, compared with CS+ unpaired processing, in nonresponders that was not evident in responders (Figure 1A). Results on baseline differences between responders and nonresponders could be replicated in the smaller subsample of 42 patients (see the online data supplement). No effects were present during familiarization or acquisition.
| All Patients (N=49)b | Patient Group | Analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Characteristic | Responders (N=25) | Nonresponders (N=24) | |||||||
| N | % | N | % | N | % | χ2 | df | p | |
| Female | 33 | 67.35 | 17 | 68.00 | 16 | 56.67 | 0.010 | 1 | 0.921 |
| Education (years) | 2.708 | 3 | 0.439 | ||||||
| 8 | 4 | 8.16 | 1 | 4.00 | 3 | 12.5 | |||
| 10 | 22 | 44.90 | 11 | 44.00 | 11 | 45.83 | |||
| 12–13 | 22 | 44.90 | 13 | 52.00 | 9 | 37.50 | |||
| No formal degree | 1 | 2.04 | 0 | 0.00 | 1 | 4.17 | |||
| Mean | SD | Mean | SD | Mean | SD | t | df | p | |
| Age (years) | 35.27 | 10.43 | 32.80 | 11.15 | 37.83 | 9.15 | 1.723 | 47 | 0.091 |
| Baseline Clinical | |||||||||
| N | % | N | % | N | % | χ2 | df | p | |
| Therapist-guided CBT arm | 28 | 57.14 | 12 | 48.00 | 16 | 66.67 | 1.742 | 1 | 0.187 |
| Comorbid depressionc | 19 | 38.78 | 9 | 36.00 | 10 | 41.67 | 0.166 | 1 | 0.684 |
| Mean | SD | Mean | SD | Mean | SD | t | df | p | |
| Number of diagnoses | 2.59 | 1.35 | 2.40 | 1.29 | 2.79 | 1.41 | 1.013 | 47 | 0.316 |
| CGI score | 5.39 | 0.67 | 5.32 | 0.69 | 5.46 | 0.66 | 0.717 | 47 | 0.477 |
| SIGH-A total score | 24.59 | 5.28 | 24.16 | 5.39 | 25.04 | 5.23 | 0.581 | 47 | 0.564 |
| Panic and Agoraphobia Scale total score | 26.75 | 8.67 | 21.93 | 7.73 | 31.78 | 6.54 | 0.221 | 47 | <0.001 |
| Anxiety Sensitivity Index total score | 31.31 | 10.00 | 32.56 | 8.28 | 30.00 | 11.56 | –0.894 | 47 | 0.376 |
| BDI-II total score | 17.35 | 8.69 | 17.44 | 9.93 | 17.25 | 7.39 | –0.076 | 44 | 0.940 |
| Posttreatment Clinical | |||||||||
| CGI score | 3.57 | 1.00 | 3.16 | 1.07 | 4.00 | 0.72 | 3.212 | 47 | 0.002 |
| SIGH-A total score | 13.10 | 6.96 | 7.96 | 3.48 | 18.46 | 5.46 | 7.897 | 39 | <0.001 |
| Panic and Agoraphobia Scale total scored | 14.20 | 8.47 | 9.19 | 5.48 | 19.64 | 7.81 | 5.324 | 39 | <0.001 |
| Anxiety Sensitivity Index total score | 16.18 | 9.17 | 13.24 | 7.81 | 19.25 | 9.63 | 2.404 | 47 | 0.020 |
| BDI-II total score | 9.47 | 7.48 | 6.56 | 5.75 | 12.50 | 7.98 | 3.000 | 47 | 0.004 |
| Neuropsychological | |||||||||
| Digit-span forward task | 7.82 | 1.88 | 8.16 | 1.86 | 7.46 | 1.86 | –1.317 | 47 | 0.194 |
| Digit-span backward task | 7.14 | 1.95 | 7.48 | 2.06 | 6.79 | 1.79 | –1.244 | 47 | 0.220 |
| Trail Making Test, part A (seconds) | 26.01 | 8.85 | 25.18 | 8.59 | 26.87 | 9.21 | 0.663 | 47 | 0.510 |
| Trail Making Test, part B (seconds) | 56.22 | 16.95 | 53.32 | 14.61 | 59.23 | 18.93 | 1.221 | 43 | 0.226 |
| Contrast and Region | Hemisphere | Voxels (Number Per Cluster) | MNI Coordinates (x, y, z) | F or t | p (Uncorrected)b |
|---|---|---|---|---|---|
| Differences at baseline | |||||
| Familiarization phase | |||||
| Main effect of groupc | |||||
| Interaction effect of group-by-CSc | |||||
| Acquisition phase | |||||
| Main effect of groupc | |||||
| Interaction effect of group-by-CSc | |||||
| Extinction phase | |||||
| Main effect of groupc | |||||
| Interaction effect of group-by-CS | |||||
| Anterior cingulate gyrusd | Right | 292 | 20, 42, 0 | 20.55 | <0.001 |
| Fusiform gyrus | Left | 199 | –42, –46, –20 | 15.42 | <0.001 |
| Middle temporal gyrus | Left | 220 | –48, –62, 12 | 14.78 | <0.001 |
| Hippocampuse | Right | 250 | 42, –18, –10 | 13.02 | <0.001 |
| Amygdala (region-of-interest analysis)f | Right | 18 | 30, 4, –26 | 13.80 | 0.012 |
| Post hoc t contrasts | |||||
| Responders (CS+ > CS–) > nonresponders (CS+ > CS–) | |||||
| Anterior cingulate gyrusd | Right | 292 | 20, 42, 0 | 4.53 | <0.001 |
| Fusiform gyrus | Left | 199 | –42, –46, –20 | 3.93 | <0.001 |
| Middle temporal gyrus | Left | 220 | –48, –62, 12 | 3.84 | <0.001 |
| Hippocampuse | Right | 250 | 42, –18, –10 | 3.61 | <0.001 |
| Amygdala (region-of-interest analysis)f | Right | 29 | 30, 4, –26 | 3.71 | 0.006 |
| Nonresponders (CS+ > CS–) > responders (CS+ > CS–)c | |||||
| Neuroplastic changes from baseline to posttreatment assessment, extinction phase | |||||
| Interaction effect of group-by-time | |||||
| Hippocampus | Right | 449 | 40, –24, –12 | 22.61 | <0.001 |
| Superior temporal gyrus | Right | 303 | 40, –44, 8 | 17.77 | <0.001 |
| Superior medial frontal gyrus | Right | 243 | 16, 46, 6 | 13.73 | <0.001 |
| Inferior frontal gyrus, triangular | Left | 321 | –50, 26, 6 | 13.49 | <0.001 |
| Precuneusg | Left | 175 | –24, –50, 2 | 13.32 | <0.001 |
| Superior medial frontal gyrus | Left | 522 | –8, 60, 20 | 12.38 | <0.001 |
| Post hoc t contrasts | |||||
| Responders: baseline > posttreatment assessmentc | |||||
| Responders: posttreatment assessment > baselinec | |||||
| Hippocampus | Right | 252 | 40, –18, –18 | 4.15 | <0.001 |
| Nonresponders: baseline > posttreatment assessment | |||||
| Superior frontal gyrus | Right | 146 | 16, 50, 30 | 3.83 | <0.001 |
| Anterior cingulate gyrush | Right | 180 | 20, 40, 10 | 3.76 | <0.001 |
| Precuneusi | Right | 202 | 28, –44, 8 | 3.49 | <0.001 |
| Inferior frontal gyrus, triangular | Left | 221 | –34, 28, 8 | 3.23 | <0.001 |
| Nonresponders: posttreatment assessment > baselinec | |||||
| Interaction effect of group-by time-by-CS | |||||
| Middle temporal gyrus | Left | 216 | –46, –64, 14 | 15.61 | <0.001 |
| Middle temporal gyrus | Left | 245 | –56, –34, –8 | 14.01 | <0.001 |
| Fusiform gyrus | Left | 189 | –28, –62, –6 | 13.32 | <0.001 |
| Hippocampus | Right | 168 | 40, –16, –14 | 11.84 | <0.001 |
| Post hoc t contrasts | |||||
| Responders: baseline > posttreatment assessment (CS+ > CS–)c | |||||
| Responders: posttreatment assessment > baseline (CS+ > CS–)c | |||||
| Nonresponders: baseline > posttreatment assessment (CS+ > CS–)c | |||||
| Nonresponders: posttreatment assessment > baseline (CS+ > CS–) | |||||
| Hippocampus | Right | 142 | 40, –16, –16 | 3.94 | <0.001 |
| Middle temporal gyrus | Left | 182 | –56, –32, –10 | 3.70 | <0.001 |

a In panel A, differences are as indicated by the interaction effect of group-by-CS (conditioned stimulus) during the extinction phase (error bars indicate the standard error of the mean). Estimated beta values from the pregenual anterior cingulate cortex (ACC), amygdala, and hippocampus cluster show that the effect is driven by enhanced activation toward the CS– (conditioned stimulus not followed by the unconditioned stimulus, safety signal) during the extinction phase in nonresponders but not in responders. In panel B, group differences in functional connectivity between the ACC and the amygdala are shown. Connectivity was analyzed across the entire time course of the conditioning paradigm. The activation cluster of the ACC served as the seed region. Results are presented using a region-of-interest approach for the amygdala (p<0.05, family-wise-error corrected). Responders and nonresponders differed in functional connectivity between these two regions, with responders showing a negative coupling between the ACC and the amygdala (error bars indicate the standard error of the means). Panel C presents classification accuracy for treatment response, using neural activation in the ACC in response to the CS– (extinction phase) as a predictor. ROC=receiver operating characteristics; AUC=area under the curve; L=left; R=right.
*p<0.05. **p<0.01. ***p<0.001.
Group differences in functional connectivity with the anterior cingulate cortex cluster as a seed region were found in the left amygdala, the right superior medial frontal and precentral gyri, and the left superior frontal and parahippocampal gyri, with responders exhibiting a negative connectivity but nonresponders exhibiting a positive connectivity. Region-of-interest analyses confirmed findings in the amygdala using a conservative statistical threshold (Table 3, Figure 1B).
| Contrast and Region | Hemisphere | Voxels (Number Per Cluster) | MNI Coordinates (x, y, z) | F or t | p (Uncorrected) |
|---|---|---|---|---|---|
| Differences at baseline (whole-brain analysis)a | |||||
| Nonresponders > responders | |||||
| Amygdalab | Left | 8 | –22, 2, –28 | 3.05 | 0.002 |
| Superior medial frontal gyrus | Right | 3 | 8, 54, 46 | 2.84 | 0.003 |
| Precentral gyrus | Right | 10 | 54, 12, 34 | 2.83 | 0.004 |
| Superior frontal gyrus | Left | 1 | –18, 48, 46 | 2.74 | 0.004 |
| Parahippocampal gyrus | Left | 1 | –22, –24, –28 | 2.71 | 0.005 |
| Neuroplastic changes from pre- to posttreatment assessment (region-of-interest analysis)c | |||||
| Main effect of group | |||||
| Amygdala | Left | 3 | –22, 0, –28 | 14.27 | 0.016 |
| Main effect of timed | |||||
| Interaction effect of group-by-timed |
The anterior cingulate cortex and hippocampus cluster yielded good classification accuracies for treatment response, with an area under the curve exceeding the value that would be expected by chance (anterior cingulate cortex: area under the curve=0.758, p=0.002; hippocampus: area under the curve=0.726, p=0.007; amygdala: area under the curve=0.563, p=0.447; anterior cingulate cortex-amygdala connectivity: area under the curve=0.642, p=0.089) (Figure 1C).
Neuroplastic Changes as a Function of Treatment Response
Demographic and clinical characteristics of the baseline and posttreatment assessment sample are presented in the online data supplement. Significant clinical improvement was present in both responders and nonresponders, albeit more pronounced in responders (Figure 2A). We observed a significant two-way interaction of group-by-time and three-way interaction of group-by-time-by-CS. Post hoc tests on the three-way interaction revealed increased activation in the right hippocampus in the nonresponder group in response to the safety stimulus at baseline, but this activation was reduced at the posttreatment assessment (contrast: nonresponder group: posttreatment assessment > baseline [CS+ > CS–]; Figure 2C). A similar response pattern was evident for the two-way interaction in the right anterior cingulate cortex cluster, which revealed reduced activity in nonresponders at the posttreatment assessment (contrast: nonresponder group: baseline > posttreatment assessment). Particular successful treatment response was characterized by enhanced activation in the right hippocampus during the processing of both the CS+ and CS– (contrast: responder group: posttreatment assessment > baseline; Figure 2B). No changes over time were observed in functional anterior cingulate cortex-amygdala connectivity (Table 3).
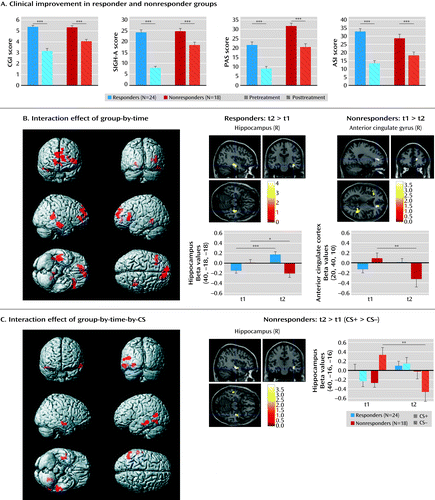
a The graphs in panel A compare measures of clinical improvement for responders (N=24) and nonresponders (N=18). CGI=Clinical Global Impressions Scale; SIGH-A=Structured Interview Guide for the Hamilton Anxiety Rating Scale; PAS=Panic and Agoraphobia Scale; ASI=Anxiety Sensitivity Index. In panel B, neuroplastic changes, as indicated by the interaction effect of group by time from baseline (t1) to posttreatment assessment (t2), are presented. Post hoc tests show that this effect is driven by increased activation in the right hippocampus in responders but decreased activation in the anterior cingulate cortex in nonresponders. In panel C, neuroplastic changes, as indicated by the interaction of group-by-time-by-CS (conditioned stimulus), are shown. This effect is driven by a reduction of activation in the right hippocampus in response to the CS– (conditioned stimulus not followed by the unconditioned stimulus, safety signal) from t1 to t2 in nonresponders. Error bars indicate the standard error of the mean.
*p<0.05. **p<0.01. ***p<0.001.
Discussion
Despite increasing evidence on the neurobiology of panic disorder (25, 26), there is a lack of knowledge about the predictive value of neural activation patterns for therapeutic outcome. We focused on neural correlates of fear conditioning and neuroplastic changes after CBT as a marker of the pathophysiology of panic disorder with agoraphobia and putative pathways of change. Patients who did not respond to treatment exhibited enhanced activation in the pregenual anterior cingulate cortex, the amygdala, and the hippocampus during safety signal processing compared with responders. These increased activations among nonresponders normalized after CBT, while treatment response was associated with an increase in hippocampal activation when processing stimulus contingencies. Nonresponse was further associated with a lack of inhibitory functional anterior cingulate cortex-amygdala coupling that did not change after CBT.
Fear conditioning has been utilized as an experimental approach to better understand pathological forms of anxiety. It has been demonstrated to correlate with activity in a neural network encompassing the amygdala, the hippocampus, and the anterior cingulate cortex in the human brain (27). Our study is one of the first, to our knowledge, to apply fear conditioning as a neurofunctional marker of treatment response in patients with panic disorder with agoraphobia. At baseline, enhanced activation in the above-described network in nonresponders was observed during the processing of stimuli that signal safety. These findings not only corroborate results of behavioral (5, 6) and neuroimaging (8) studies of altered safety signal processing in persons with panic disorder, but also show that activation of a neural network signaling fear in response to harmless stimuli is associated with treatment nonresponse. This may imply that patients with a detection bias toward threat, not differentiating safe versus unsafe contexts, do not sufficiently benefit from exposure-based treatment. Receiver operating characteristic curve analyses yielded good classification accuracy of neural activation patterns for treatment response, although the predictive value must be tested in a second, independent sample.
Our findings also indicated, however, that the dysfunctional baseline activations in patients who did not respond to treatment resolved after CBT, as indicated by reduced activity in the right hippocampus and anterior cingulate cortex. Although these patients were classified as nonresponders, they exhibited significant symptom reduction, indicating that they improved after CBT but not as much as patients who were classified as responders. Dysfunctional predispositions, as reflected by a neural bias toward threat detection, might disadvantage certain patients in therapy. In contrast, treatment response was associated with enhanced hippocampal activation when processing stimulus contingencies of both the CS+ and CS–. Brain lesion studies have emphasized the role of the hippocampus in conscious contingency awareness (28) during fear conditioning. Since exposure therapy has been reported to involve strong conscious components (29), we assume not only that sustained behavioral and neural changes after CBT require the unlearning of emotional responses, but also that this is most effective when contingencies between contexts, stimuli, and individual experiences are consciously learned and reappraised, as may be reflected by hippocampal involvement.
In a previous fMRI study (9), changes of brain activation patterns in nine patients with panic disorder following short-term inpatient psychodynamic treatment were investigated using an emotional linguistic go/no-go task. Increased activation in the hippocampus and amygdala and low activation in the prefrontal cortex normalized after symptom improvement, but it is unclear whether these activation patterns can be generalized to the process of fear conditioning. Previous analyses of a subset of the present sample investigated the overall effect of CBT using a fear-conditioning task, comparing patients with healthy subjects. Enhanced activation in the left inferior frontal gyrus attenuated over time after CBT (3). In line with a function of the inferior frontal gyrus in cognitive appraisal of negative emotions and threat (30), results were interpreted as a reduction of negative cognitions following treatment. Supplementing these global effects of CBT, our data suggest that the amount of treatment success is modulated by additional neural circuits, such as medial prefrontal-limbic networks. Although aberrant activation in the amygdala could be identified as a baseline characteristic of nonresponse, we did not observe treatment-related changes in this structure. Findings evidencing general hyperactivation in the amygdala in persons with panic disorder have been inconsistent (31) and may apply more to state than trait characteristics (25).
Anterior cingulate cortex/medial prefrontal cortex-amygdala interactions have been implicated in fear extinction, emotion regulation, and trait anxiety (32, 33). Successful fear extinction has been ascribed to inhibitory top-down modulation of the amygdala through medial prefrontal cortex inputs (34), and functional connectivity between the amygdala and ventromedial prefrontal cortex has been observed during fear extinction and emotion regulation in humans (35). Pezawas et al. (32) reported a functional distinction between pregenual and subgenual components of the anterior cingulate cortex, with the former being negatively coupled with the amygdala, while the latter exhibited a positive coupling. The anterior cingulate cortex seed region we used in this study is located in the pregenual area, thus corroborating the notion of an inhibitory relationship to the amygdala. As shown in our results, this brain circuit is functionally relevant for treatment response in exposure-based CBT: responders were already characterized by a relatively higher inhibitory connectivity in this circuit before treatment. The chance to benefit from exposure in which extinction learning is conveyed through medial prefrontal cortex/anterior cingulate cortex-amygdala interactions may be increased in those patients who already have a relatively strong inhibitory coupling between these structures before therapy. Replicating previous findings of stable fronto-limbic connectivity in this sample (3), we did not observe significant changes over time. This may indicate that the observed pattern of connectivity represents either a vulnerability to or a trait factor for panic disorder with agoraphobia, a hypothesis that could be tested in high-risk samples. Alternatively, changes in functional connectivity may require therapeutic interventions of a longer duration than we used in this study.
There are several limitations to this study. About one-third of patients initially scanned could not be considered for the analysis. The subsample examined was, however, clinically comparable to both patients who dropped out of the fMRI study and those in the non-fMRI sample. Comorbid diagnoses were not excluded per se, since presence of comorbid depression or anxiety conforms to what is usually seen in practice and thus may improve the external validity of the sample. The number of diagnoses, particularly depressive disorders, was comparable between responders and nonresponders. We included depression scores as a covariate in the model to account for psychopathology that was not specific to panic disorder with agoraphobia. Autonomic indices of fear conditioning were not available for our sample because of site-specific technical restrictions, but a pilot study indicated successful fear conditioning during the task (21). No further markers are available to support the hypothesis of altered safety signal processing. Finally, the study design is lacking an extinction recall phase. Main effects were observed in the extinction learning phase but not in the acquisition phase, making it difficult to distinguish between processes related to the recall of the conditioned response and the gradual induction of extinction. Including a familiarization phase that preceded the acquisition phase most likely induced latent inhibition (an effect in which preconditioning exposure to the CS delays subsequent conditioning), thus possibly shifting the recall of conditioned responses further into the extinction phase. Future studies should more closely address extinction deficits in this patient population (7) using tasks that allow for a separate analysis of extinction learning and recall.
In summary, this study identified a brain network associated with treatment response in patients with panic disorder with agoraphobia. Altered safety signal processing and impaired inhibitory anterior cingulate cortex-amygdala coupling that will augment, rather than down-regulate, fear-circuit reactivity may represent an important baseline characteristic that predisposes a subgroup of patients to obtain less benefit from CBT. Our findings can guide future add-on approaches, such as repetitive transcranial magnetic simulation (36) or neurofeedback (37), to purposefully influence “disadvantageous” brain activity in patients who do not sufficiently respond to CBT. While this dysfunctional baseline pattern partly resolved after CBT, treatment response was characterized by neuroplastic change in the hippocampus, possibly indicating conscious encoding strategies. These findings may not only contribute to a better understanding of how neurofunctional predispositions interact with behavioral treatments in these patients, but they may also enlarge our knowledge about the pathways by which successful CBT is conveyed.
1 : Empirically supported treatments for panic disorder. Psychiatr Clin North Am 2009; 32:593–610Crossref, Medline, Google Scholar
2 : A modern learning theory perspective on the etiology of panic disorder. Psychol Rev 2001; 108:4–32Crossref, Medline, Google Scholar
3 : Effect of cognitive-behavioral therapy on neural correlates of fear conditioning in panic disorder. Biol Psychiatry 2013; 73:93–101Crossref, Medline, Google Scholar
4 : Functional imaging in anxiety disorders. Verhaltenstherapie 2009; 19:78–85Crossref, Google Scholar
5 : Impaired discriminative fear-conditioning resulting from elevated fear responding to learned safety cues among individuals with panic disorder. Behav Res Ther 2009; 47:111–118Crossref, Medline, Google Scholar
6 : Overgeneralization of conditioned fear as a pathogenic marker of panic disorder. Am J Psychiatry 2010; 167:47–55Link, Google Scholar
7 : Fear conditioning in panic disorder: enhanced resistance to extinction. J Abnorm Psychol 2007; 116:612–617Crossref, Medline, Google Scholar
8 : Differential activity of subgenual cingulate and brainstem in panic disorder and PTSD. J Anxiety Disord 2011; 25:251–257Crossref, Medline, Google Scholar
9 : Changes of brain activation pre- post short-term psychodynamic inpatient psychotherapy: an fMRI study of panic disorder patients. Psychiatry Res 2010; 184:96–104Crossref, Medline, Google Scholar
10 : The change of regional brain metabolism (18FDG PET) in panic disorder during the treatment with cognitive behavioral therapy or antidepressants. Neuroendocrinol Lett 2004; 25:340–348Medline, Google Scholar
11 : Changes in cerebral glucose utilization in patients with panic disorder treated with cognitive-behavioral therapy. Neuroimage 2006; 33:218–226Crossref, Medline, Google Scholar
12 : Changes in cerebral cortex and limbic brain functions after short-term paroxetine treatment in panic disorder: an [F]FDG-PET pilot study. Psychiatry Investig 2010; 7:215–219Crossref, Medline, Google Scholar
13 : Psychological treatment for panic disorder with agoraphobia: a randomized controlled trial to examine the role of therapist-guided exposure in situ in CBT. J Consult Clin Psychol 2011; 79:406–420Crossref, Medline, Google Scholar
14 : Reliability and validity of a structured interview guide for the Hamilton Anxiety Rating Scale (SIGH-A). Depress Anxiety 2001; 13:166–178Crossref, Medline, Google Scholar
15 : MAOA and mechanisms of panic disorder revisited: from bench to molecular psychotherapy. Mol Psychiatry (Epub ahead of print, Jan 15, 2013Google Scholar
16 : ECDEU Assessment Manual for Psychopharmacology, Revised. Rockville, Md, U S National Institute of Health, Psychopharmacology Research Branch, 1976Google Scholar
17 : Panic and Agoraphobia Scale (PAS). Ashland, Ohio, Hogrefe and Huber Publishers, 1999Google Scholar
18 : Anxiety sensitivity, anxiety frequency and the prediction of fearfulness. Behav Res Ther 1986; 24:1–8Crossref, Medline, Google Scholar
19 : Beck Depression Inventory, 2nd ed. San Antonio, Tex, Psychological Corporation, 1996Google Scholar
20 : DIA-X Interview. Frankfurt, Germany, Swets and Zeitlinger, 1997Google Scholar
21 : Neural correlates of aversive conditioning: development of a functional imaging paradigm for the investigation of anxiety disorders. Eur Arch Psychiatry Clin Neurosci 2010; 260:443–453Crossref, Medline, Google Scholar
22 ;
23 : Distinct prefrontal cortex activity associated with item memory and source memory for visual shapes. Brain Res Cogn Brain Res 2003; 17:75–82Crossref, Medline, Google Scholar
24 : An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage 2003; 19:1233–1239Crossref, Medline, Google Scholar
25 : Revise the revised? New dimensions of the neuroanatomical hypothesis of panic disorder. J Neural Transm 2013; 120:3–29Crossref, Medline, Google Scholar
26 : Current findings of fMRI in panic disorder: contributions for the fear neurocircuitry and CBT effects. Expert Rev Neurother 2010; 10:291–303Crossref, Medline, Google Scholar
27 : Human fear conditioning and extinction in neuroimaging: a systematic review. PLoS ONE 2009; 4:e5865Crossref, Medline, Google Scholar
28 : Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science 1995; 269:1115–1118Crossref, Medline, Google Scholar
29 : Cognitive processes during fear acquisition and extinction in animals and humans: implications for exposure therapy of anxiety disorders. Clin Psychol Rev 2008; 28:199–210Crossref, Medline, Google Scholar
30 : Neural mechanisms underlying selective attention to threat. Ann N Y Acad Sci 2008; 1129:141–152Crossref, Medline, Google Scholar
31 : Amygdala responses to masked and low spatial frequency fearful faces: a preliminary fMRI study in panic disorder. Psychiatry Res 2012; 203:159–165Crossref, Medline, Google Scholar
32 : 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 2005; 8:828–834Crossref, Medline, Google Scholar
33 : The structural and functional connectivity of the amygdala: from normal emotion to pathological anxiety. Behav Brain Res 2011; 223:403–410Crossref, Medline, Google Scholar
34 : Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 2002; 420:70–74Crossref, Medline, Google Scholar
35 : Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron 2008; 59:829–838Crossref, Medline, Google Scholar
36 : Randomized sham controlled trial of repetitive transcranial magnetic stimulation to the dorsolateral prefrontal cortex for the treatment of panic disorder with comorbid major depression. J Affect Disord 2013; 144:153–159Crossref, Medline, Google Scholar
37 : Acquired self-control of insula cortex modulates emotion recognition and brain network connectivity in schizophrenia. Hum Brain Mapp 2013; 34:200–212Crossref, Medline, Google Scholar

