
Variants in Nicotinic Receptors and Risk for Nicotine Dependence
Abstract
Objective: A recent study provisionally identified numerous genetic variants as risk factors for the transition from smoking to the development of nicotine dependence, including an amino acid change in the α5 nicotinic cholinergic receptor ( CHRNA5 ). The purpose of this study was to replicate these findings in an independent data set and more thoroughly investigate the role of genetic variation in the cluster of physically linked nicotinic receptors, CHRNA5-CHRNA3-CHRNB4 , and the risk of smoking. Method: Individuals from 219 European American families (N=2,284) were genotyped across this gene cluster to test the genetic association with smoking. The frequency of the amino acid variant (rs16969968) was studied in 995 individuals from diverse ethnic populations. In vitro studies were performed to directly test whether the amino acid variant in the CHRNA5 influences receptor function. Results: A genetic variant marking an amino acid change showed association with the smoking phenotype (p=0.007). This variant is within a highly conserved region across nonhuman species, but its frequency varied across human populations (0% in African populations to 37% in European populations). Furthermore, functional studies demonstrated that the risk allele decreased response to a nicotine agonist. A second independent finding was seen at rs578776 (p=0.003), and the functional significance of this association remains unknown. Conclusions: This study confirms that at least two independent variants in this nicotinic receptor gene cluster contribute to the development of habitual smoking in some populations, and it underscores the importance of multiple genetic variants contributing to the development of common diseases in various populations.
Cigarette smoking is a major public health problem that contributes to nearly 5 million deaths every year worldwide (1) . Despite knowledge of the adverse health effects, 45 million adults in the United States smoke, and about half of these individuals are dependent on nicotine (2 , 3) . Nicotine is the component in cigarettes that is responsible for the maintenance of smoking, and the physiological effects of this drug are mediated largely through the neuronal nicotinic acetylcholine receptors (nAChRs) (4) .
Our group recently completed genome-wide association and candidate gene studies of nicotine dependence based on the contrast between nicotine-dependent smokers and smokers who used at least 100 cigarettes in their lifetime but never developed any symptoms of dependence (5 , 6) . These genetic studies focused on the transition from smoking to the development of nicotine dependence. Intriguing findings for further follow-up included the identification of a strong association of nicotine dependence with genetic polymorphisms in the nicotinic receptor gene cluster, α5-α3-β4, on chromosome 15, which included a variant that results in an amino acid change (aspartic acid [D] to asparagine [N]) in the α5 neuronal nicotinic acetylcholine receptor subunit ( CHRNA5 ). There was also evidence of a second distinct finding in this cluster marked by rs578776 (pairwise r 2 <0.2 with rs16969968) in the α5-α3-β4 gene cluster.
Neuronal nicotinic receptors are pentameric ligand-gated cation channels that are expressed in the central and peripheral nervous systems and are composed of different combinations of primarily α and β subunits (see reference 7 for a review). The α5 subunit participates in multiple nicotinic receptor subtypes (8 – 14) , including an α4β2α5 subtype that contributes to nicotine-stimulated dopamine release in the striatum, a region that is involved in the reward pathway and is crucial to the development of substance dependence (15 , 16) . The α3 subunit has limited distribution in the brain and forms α3β2 and α3β4 receptors (7) .
The purpose of this study was to further define the genetic contribution of these findings to smoking in families affected with alcoholism. This involved testing whether the original observations could be replicated and fine-mapped in an independent data set, examining the frequency of the variant resulting in the amino acid change in diverse populations and performing a functional study to determine whether the amino acid substitution changed receptor function.
Method
Collaborative Study on the Genetics of Alcoholism
The Collaborative Study on the Genetics of Alcoholism (COGA), a genetic study of alcohol dependence, had high rates of smoking and allowed for the genetic study of habitual smoking and light smoking contrast groups (17) . The institutional review boards of all participating institutions approved the study. All study subjects provided written informed consent after receiving a complete description of the study.
Alcohol-dependent probands were identified through inpatient or outpatient chemical dependency treatment programs. The Semi-Structured Assessment for the Genetics of Alcoholism (18) was administered to probands and their relatives. Although a diagnostic assessment of nicotine dependence was not administered to all subjects, smoking history was collected and proxies for nicotine-dependent and nondependent status were developed. Case status was classified by habitual smoking, defined as ever smoking at least one pack (20 cigarettes) daily for 6 months or more. Subjects were classified as unaffected if they had smoked at least 100 cigarettes in their lifetime or had smoked daily for 1 month or more but never smoked more than 10 cigarettes daily (19) . In a subset of subjects who were also assessed with the Fagerström Test for Nicotine Dependence (20) , 78% of habitual smokers were categorized as nicotine dependent when a score of 4 or more was used as the cutoff (the scale runs from 0 to 10, with higher scores indicating greater dependence), and 73% of light smokers were not nicotine dependent (see Figure S1 in the data supplement that accompanies the online edition of this article). Those who never smoked or did not meet the affected or unaffected status were considered “unknown” phenotypically in the genetic analyses.
Each family that participated in the genetic phase of this study included an alcohol-dependent proband and at least two first-degree relatives with alcohol dependence. A total of 262 families including 2,309 individuals were selected for the genetic study (21 , 22) . The 219 pedigrees of European descent, with 2,284 genotyped individuals including 955 habitual smokers and 281 light smokers, were analyzed.
Human Diversity Cell Line Panel
The Human Genome Diversity Cell Line Panel (from the Human Genome Diversity Project [HGDP] and the Centre d’Etude du Polymorphisme Humain [CEPH]) is a resource of DNA from individuals from different world populations that is banked at the Foundation Jean Dausset-CEPH in Paris (23) . These samples were collected as part of the HGDP to provide DNA for studies of sequence diversity and history of human populations. Information includes the population and geographic origin of the samples. Genetic structure analysis on these human populations identified six main genetic clusters, five of which correspond to major geographic regions, and subclusters that often correspond to individual populations (24) . Informed consent was obtained from all subjects.
SNP Assays
The dbSNP database established by the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/SNP/) was used to identify single nucleotide polymorphisms (SNPs) within and flanking the α5-α3-β4 gene cluster. Sequenom MassArray technology (http://www.sequenom.com), homogeneous MassEXTEND (hME), or iPLEX assays were used for genotyping. A detailed protocol has been described elsewhere (25) . The primer sequences used for genotyping are listed in Table S1 in the online data supplement. For the 22-base-pair (bp) insertion/deletion polymorphism (rs3841324), polymerase-chain-reaction (PCR) primers (forward primer 5′-AAAAGGAACAAGGCGAGGATTG-3′; reverse primer 5′-GAGTGTGAGTCGTGAGACAAAACG-3′) were selected using the MacVector 6.5.3 software package (Accelrys, Inc., San Diego) to yield a 166-bp or 188-bp genomic fragment containing the SNP rs3841324. The amino acid change coding SNP in exon 5 of the CHRNA5 gene, rs16969968, was genotyped using a restriction fragment length polymorphism assay. PCR products were generated with forward primer 5′-CGCCTTTGGTCCGCAAGATA-3′ and reverse primer 5′-TGCTGATGGGGGAAGTGGAG-3′ and then digested with Taq1 restriction enzyme. Genotypes of rs3841324 and rs16969968 were detected by electrophoresis on 2% agarose gel.
Statistical Analyses
Linkage disequilibrium between markers was computed using the software program Haploview (26) . The family-based association test was used to examine the association between genetic variants and habitual smoking (27) . The significant covariates, sex and age, were incorporated in the model. There was no correction for multiple testing.
Functional Studies of CHRNA5 Genetic Variant
Cell culture
HEK293T cells were maintained at 37°C in a humidified 5% CO 2 environment in Dulbecco’s modified Eagle’s medium (high glucose, no pyruvate), 10% heat-inactivated fetal bovine serum, and antibiotic/antimycotic (100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B). Culture reagents were purchased from either Biowhittaker (East Rutherford, N.J.) or Invitrogen (Carlsbad, Calif.).
Measurement of intracellular calcium
Agonist (epibatidine)-evoked changes in intracellular calcium were performed using an aequorin-based luminescence assay as previously described (28) . HEK293T cells were seeded onto six-well plates (1.5×10 6 cells/well) and were transfected the following day with plasmids (0.25 μg/well for each plasmid) containing a human codon-optimized aequorin cDNA (29) , the mouse α4 and β2 cDNAs, and either the wild-type mouse a5 cDNA (D398) or a mouse α5 cDNA in which D398 was mutated to N398. Transfection was performed using either the LipofectAmine Plus Reagent (Invitrogen) or FuGENE HD transfection reagent (Roche Applied Science, Indianapolis) as recommended by the manufacturers. Approximately 48 hours after transfection, culture medium was replaced with Dulbecco’s modified Eagle’s medium plus 0.1% fetal bovine serum and 2.5 μm coelenterazine-hcp (Invitrogen), and the cells were incubated for 3 hours at 37°C in a humidified 5% CO 2 incubator. After the coelenterazine incubation, cells were gently aspirated from the culture dishes and transferred to 2-ml tubes. The cells were then pelleted by centrifugation at 4°C for 5 minutes at 800 g; the supernatant was discarded, and the cells were resuspended in 1× assay buffer (Hank’s Balanced Salt Solution [Cambrex, East Rutherford, N.J.], supplemented to 10 mM CaCl 2 ) and incubated for 1 hour at 4°C prior to initiating the assay. The sample size was 12 for each nAChR variant (12 separate transfections per variant from three independent experiments).
For the epibatidine concentration response curves, 50 μL of cells were added to each well of a 96-well opaque white plate and placed in a Victor 3 V plate reader (PerkinElmer, Waltham, Mass.). After a 1-second baseline read, 50 μL of epibatidine was injected onto each sample and luminescence (in lux) was recorded at 0.2-second intervals for 20 seconds immediately after the addition of agonist. At the completion of the agonist stimulation, 100 μL of a solution containing 0.1% Triton X-100 and 100 mM CaCl 2 was injected into each well, and luminescence was recorded for 5 seconds at 0.1-second intervals. To control for differences in cell number per well as well as variation in transfection efficiency and coelenterazine loading, agonist responses were normalized by dividing the maximal peak value for the agonist-stimulated luminescence (L) by the total peak luminescence value (L max ) (maximal peak agonist-stimulated luminescence + maximal peak luminescence resulting from cell lysis in the presence of high calcium).
The half-maximal effective concentration (EC 50 ) and maximal response values for the concentration response curves were calculated using a four-parameter logistic equation in the Graphpad Prism 3.0 software package (Graphpad Software, San Diego). Concentration response curves for the two nAChR populations were evaluated using two-way analysis of variance (ANOVA) for epibatidine concentration and receptor variant. Maximal response and EC 50 values between the α4β2α5D398 and α4β2α5N398 were compared using Student’s t test (two-tailed). Verification that the a5 subunit was being incorporated into functional α4β2α5 nAChRs was established using the reporter mutation approach as described by others (28 , 30 – 33) (data not shown).
[ 125 I]-Epibatidine binding
Membrane fractions were prepared from samples as previously described (34) , except that a 15-minute incubation at 37°C with 50 μg/ml DNase was performed prior to the first centrifugation. The binding of [ 125 I]-epibatidine to the membrane fractions was performed essentially as described previously (34) in a 30-μL reaction that included binding buffer (118 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl 2 , 1.2 mM Mg 2 SO 4 , 20 mM HEPES [pH 7.5]), and 200 pM [ 125 I]-epibatidine. Nonspecific binding was determined by the inclusion of 10 mmol/liter cytisine in the reaction. Ligand binding was performed with an amount of homogenate that did not produce ligand depletion. Homogenate protein levels were determined by the method described by Lowry et al. (35) . Differences in binding were assessed using Student’s t test (two-tailed).
Western blot immunochemistry
Unused samples from the aequorin assay were lysed in tris-buffered saline (25 mM tris [pH=7.4], 150 mM NaCl), 1% triton X-100, 1 mM EDTA, and protease inhibitor cocktail (Sigma, St. Louis). Equal volumes of sample were denatured in Laemmli buffer (Pierce Chemical Co., Rockford, Ill.), run on an SDS-PAGE gel, and transferred to a polyvinylidene fluoride membrane as described previously (36) . The membrane was incubated with monoclonal antibody 268 at a concentration of 1 μg/ml followed by a horseradish peroxidase (HRP)-conjugated goat-anti rat antibody (Pierce Chemical Co.) at a dilution of 1:5000. The membrane subsequently was incubated with ECL Advance (an enhanced chemiluminescent HRP substrate [GE Healthcare, Piscataway, N.J.]), and the resulting image was captured using an Image Station 2000R (Eastman Kodak, New Haven, Conn.). Intensity levels for the α5 subunit were determined using the Kodak 1D Image Analysis software package (Carestream Health, Rochester, N.Y.). Differences in protein levels were assessed using Student’s t test (two-tailed).
Results
A careful examination of the linkage disequilibrium pattern across the gene cluster revealed evidence of two distinct findings of genetic association with habitual smoking versus light smoking in the CHRNA5-CHRNA3-CHRNB4 gene cluster. The nonsynonymous coding SNP of the CHRNA5 gene, rs16969968 (p=0.007), was associated with habitual smoking. Other SNPs that were highly correlated with rs16969968 (rs2036527, rs17486278, rs1051730, rs17487223, r 2 >0.79; see Figure S2 in the online data supplement) were also associated, with p values ranging from 0.020 to 0.086 ( Table 1 ). This SNP cluster, which spans the three genes CHRNA5-CHRNA3-CHRNB4 , most likely represents one group of correlated associated genetic variants.

A second finding of association in this gene cluster, at rs578776, was statistically independent, with a low correlation with rs16969968 (r 2 <0.15) (see Table 1 and Figure S2). Between rs16969968 (highlighted in blue in Table 1 ) and rs578776 (highlighted in yellow in Table 1 ), the absolute value of D′ is high (D′=1) but in repulsion phase. The minor allele at locus rs16969968 (allele A) is on the same chromosome as the common allele at locus rs578776 (allele C), which results in the very low r 2 . Several SNPs that were moderately correlated with rs578776 (r 2 values ranging from 0.60 to 0.76) and that were significant in the previous study (6) were not associated in this family-based analysis.
The finding of the amino acid change associated with nicotine dependence at rs16969968 was further examined in biological studies. Using sequence data from public databases, the protein sequences for CHRNA5 homologues were aligned to determine conservation in the region surrounding codon 398 in divergent species. The aspartic acid residue at amino acid position 398 was completely conserved from human to chicken, suggesting that it has functional importance (see Figure 1 ).

To assess the frequency of the minor allele (A) at rs16969968 across multiple populations, this SNP was successfully typed in the Human Genome Diversity Cell Line Panel, which included 995 individuals representing 39 different populations (23) . In populations of European and Middle Eastern origin, the frequency of the A allele was 37%–43%. The A nucleotide was not detected or was uncommon in African, East Asian, and Native American populations ( Figure 2 ; see also Table S2 in the online data supplement).
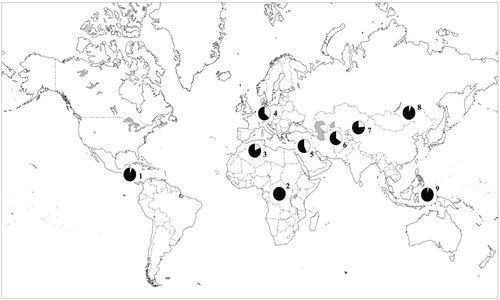
a The frequency of the A allele is the white segment of the circles in the figure. Populations were grouped together on the basis of their genetic structures as reported by Rosenberg et al. (24). Geographic regions: 1. America; 2. Africa; 3. North Africa; 4. Europe; 5. Middle East; 6. Central/South Asia; 7. Central/South Asia; 8. East Asia; 9. Oceania.
To establish whether the D398N polymorphism altered receptor function, nicotinic agonist-evoked changes in intracellular calcium were measured from HEK293T cells that heterologously expressed either α4β2α5D398 or α4β2α5N398 nAChRs. Receptor expression and α5 protein levels also were determined for each α5 variant. Two-way ANOVA indicated that the concentration response curves for the nicotinic agonist epibatidine were significantly different between the α4β2α5N398 and α4β2α5D398 nAChR variants (p<0.0001) ( Figure 3 ). This difference in concentration-response curves and maximal response to agonist was not due to a shift in sensitivity to activation by epibatidine between the nAChR variants, as their EC 50 values did not differ significantly (α4β2α5D398 EC 50 =14.7 nM [SEM=2.4]; α4β2α5N398 EC 50 =22.0 nM [SEM=3.5]; p=0.09). However, the maximal response to agonist was found to be more than two times higher for the α4β2α5D398 nAChR variant relative to the α4β2α5N398 nAChR variant (0.044 [SEM=0.002] and 0.023 [SEM=0.002], respectively; p<0.0001). In contrast, the two variant nAChRs did not differ in expression, nor were the two isoforms of the α5 subunit differentially expressed (see Figure 3 ). The sum of these data indicates that the variant forms of the α5 subunit alter receptor function without affecting receptor expression.

a Data shown in all graphs are mean values with SEMs indicated by vertical bars. Top left: Concentration-response curves for epibatidine-evoked changes in intracellular calcium were measured from HEK293T cells transfected with plasmids containing a calcium-sensing aequorin cDNA, α4 and β2 cDNAs, and either α5D398 or α5N398 cDNA (see Method section for explanation of L/L max ). Two-way analysis of variance indicated that the response curves for each nAChR variant were significantly different (N=12 per variant, p<0.0001). However, EC 50 values were not altered by the variant forms of α5 (N=12, p=0.098). Upper right: nAChRs possessing the D398 variant of α5 also exhibited a significantly greater maximal response to epibatidine as compared with nAChRs containing the N398 variant (N=12 per variant, p<0.0001). Lower left: [ 125 I] epibatidine binding on membranes prepared from cells used for the aequorin assay indicates that there is no difference in the expression of the receptors when either the D398 or N398 variant of α5 is cotransfected with α4 and β2 (N=12, p=0.375). Lower right: Western blot analysis demonstrates that the variant forms of α5 do not differ in expression (N=4 per variant, p=0.78). Lower right, inset: sample Western blot from α4β2α5D398 (D398), α4β2α5N398 (N398), and α4β2 (-) transfected cells.
Discussion
This study validates the importance of genetic variants within the α5-α3-β4 nicotinic acetylcholine receptor gene cluster that contribute to the risk of a light smoker transitioning to heavy smoking and sheds light on a potential biological mechanism. There are two distinct genetic associations—one marked by rs16969968, which results in an amino acid change in the α5 nicotinic cholinergic receptor (CHRNA5) , and a second marked by an SNP rs578776 in the 3′ untranslated region of the α3 nicotinic cholinergic receptor (CHRNA3) . Because the r 2 between these two variants is low (0.15), the statistical significance at both cannot be explained solely by the linkage disequilibrium between them. Therefore, these data imply that two distinct loci in this region alter the risk for nicotine dependence and habitual smoking. These findings confirm our previous results in a case-control series studying nicotine dependence (6) and the recent work by Berrettini et al. examining heavy smoking (37) .
In these three genetic studies demonstrating the associations, there are important differences in the definition of phenotypes, recruitment procedures, and analytic methods. The COGA sample was recruited as a high-risk population for alcoholism, and a broader smoking phenotype was used: habitual smoking, defined as smoking 20 cigarettes a day for 6 months or more, and light smoking, defined as smoking 10 cigarettes or fewer per day. A family-based analytic design for association was used, which is less likely to be biased by population stratification. The previous genetic study sample our group studied was recruited from the community, the phenotypic status was defined by the Fagerström Test for Nicotine Dependence, and a case-control design of unrelated individuals was used (5 , 6 , 20) . Similar SNPs were analyzed in these two studies. The recent publication by Berrettini et al. (37) was based on genetic studies of heart disease and other common illnesses in a population-based sample of 15,000 people. Smoking status was collected, and a quantitative phenotype defined by number of cigarettes smoked per day was studied in a secondary genetic analysis. Although different genetic variants were tested in the Berrettini et al. sample, the associated SNPs are highly correlated (in linkage disequilibrium) with the variants in our studies. The consistency of results across three different studies shows that these genetic findings are robust across populations, phenotypic classification systems, and analytic methods.
Three other research groups recently published strong evidence of genetic association of the α5-α3-β4 nicotinic acetylcholine receptor gene cluster on chromosome 15 with lung cancer (38 – 40) . The findings highlighted in these studies are highly correlated with the genetic variant that results in the amino acid change in the a5 nicotinic acetylcholine receptor gene. However, the three groups differed in their interpretation of whether this genetic association with lung cancer acts through the indirect effect of smoking or whether this variant also directly increases the vulnerability to lung cancer.
When a genetic association is found, it represents association with not only the tested variants but all genetic variants (tested and untested) that are highly correlated. Identifying the variant that causes functional changes requires biological investigations. We have focused on the amino acid change in the a5 nicotinic cholinergic receptor for further studies.
The aspartic acid at position 398 in CHRNA5 in humans occurs at a residue that is otherwise invariant across vertebrate species. Frogs, chickens, rodents, cattle, and nonhuman primates all possess an aspartic acid residue at this location. In humans, the amino acid may be either an aspartic acid, which is the predominant residue at this position, or asparagine. The α5 nicotinic subunit is not involved in receptor binding in vivo (7) , and this variant is located in the cytoplasmic loop between transmembrane domains.
Evidence that the amino acid change is functionally relevant is supported by the fact that, in vitro, α4β2α5 nicotinic receptors with the aspartic acid variant (D398) exhibited a greater maximal response to a nicotinic agonist than did α4β2α5 nicotinic receptors with the asparagine amino acid substitution (N398). Because the allele that codes for asparagine is associated with increased risk for developing nicotine dependence, and nicotinic receptors containing the α5 subunit with this amino acid (N398) exhibit reduced function in vitro, reduced function of α4β2α5 nicotinic receptors may lead to an elevated risk for developing nicotine dependence. The observation that decreased nAChR function is associated with increased risk for nicotine dependence is consistent with the observation that individuals who are extensive metabolizers of nicotine (reduced receptor activation per cigarette) are at increased risk for nicotine dependence (41 , 42) . We believe that this combined evidence of high conservation across species and biological change in receptor function supports the amino acid variant in the α5 nicotinic receptor as a causative biological factor that alters the risk of nicotine dependence, although we cannot definitively rule out the other correlated SNPs across the three-gene cluster.
The α4β2α5-containing nicotinic receptors are expressed on dopaminergic neurons in the striatum (16) , where they modulate nicotine-stimulated dopamine release (15) . In addition, α4β2α5 nicotinic receptors are also found on γ-aminobutyric acid (GABA)-ergic neurons in the striatum and ventral tegmental area (43) . This region of the brain is associated with the reward pathway, and the neurotransmitter dopamine plays a crucial role in the development of dependence. Individuals with reduced α4β2α5 cholinergic receptor activity may require greater amounts of nicotine to achieve the same activation of the dopaminergic pathway. Alternatively, reduced activity of the receptor complex on GABA-ergic neurons may lead to increased dopaminergic activity in response to nicotine. How the altered receptor activity caused by the CHRNA5 amino acid change modifies liability to nicotine dependence via the reward system in response to nicotine requires further study.
The “at risk” allele differs dramatically across human populations. It is predominantly seen in populations of European and Middle Eastern descent and is uncommon or nonexistent in populations of African, Asian, or American origin. Interestingly, African Americans have a lower prevalence of nicotine dependence than European Americans (44 , 45) , and this may be explained in part by the low prevalence of this risk allele in populations of African descent.
Less information is available regarding the second independent finding marked by the genetic variant rs578776 in this gene cluster. This SNP, rs578776, is located in the 3′ untranslated region of the CHRNA3 gene. The 3′ untranslated regions contain regulatory sequences, and we can speculate that this SNP is a putative functional variant. It is important also to note that there are correlated SNPs with rs578776 in CHRNA5 and CHRNA3 , and the localization of the functional alleles may be in either gene. Further experiments are needed to identify the potential functional variants and the biological mechanisms.
In summary, there are at least two independent genetic variants in the CHRNA5-CHRNA3-CHRNB4 gene cluster on chromosome 15 that are highly associated with smoking behaviors, and we have extended our work to identify a potential biological mechanism for one of the findings. This study provides strong evidence that an amino acid change in the α5 nicotinic receptor, which is highly conserved across species, results in a functional change that is associated with a smoker’s risk of transitioning from nondependence (light smoking) to dependence (habitual smoking) on nicotine. This variant is common in populations of European and Middle Eastern descent and increases the risk of developing nicotine dependence, but it is rare in populations of African, American, and Asian descent. Intriguingly, three recent papers demonstrated that this genetic locus also contributes to the risk of developing lung cancer. A second distinct finding in this gene cluster is also seen, although further study is needed to localize the potential functional allele and to determine whether the variant affects the expression or function of the CHRNA5 or CHRNA3 gene. These converging genetic associations and biological data support the importance of CHRNA5 and potentially CHRNA3 in the development of nicotine dependence and highlight the pharmacogenetic response to nicotine that increases the susceptibility to dependence. These findings may help predict response to pharmacologic therapies, such as varenicline and nicotine replacement, for smokers who attempt to quit, and it may shed important light on the biological mechanisms that contribute to lung cancer.
1. World Health Organization: The Facts About Smoking and Health, May 30, 2006. http://www.wpro.who.int/media_centre/fact_sheets/fs_20060530.htmGoogle Scholar
2. Grant BF, Hasin DS, Chou SP, Stinson FS, Dawson DA: Nicotine dependence and psychiatric disorders in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry 2004; 61:1107–1115Google Scholar
3. Centers for Disease Control and Prevention (CDC): Tobacco use among adults: United States, 2005. MMWR Morb Mortal Wkly Rep 2006; 55:1145–1148Google Scholar
4. Benowitz NL: Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol 1996; 36:597–613Google Scholar
5. Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, Swan GE, Rutter J, Bertelsen S, Fox L, Fugman D, Goate AM, Hinrichs AL, Konvicka K, Martin NG, Montgomery GW, Saccone NL, Saccone SF, Wang JC, Chase GA, Rice JP, Ballinger DG: Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 2007; 16:24–35Google Scholar
6. Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP, Bierut LJ: Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 2007; 16:36–49Google Scholar
7. Gotti C, Zoli M, Clementi F: Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 2006; 27:482–491Google Scholar
8. Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L: Functional contributions of α5 subunit to neuronal acetylcholine receptor channels. Nature 1996; 380:347–351Google Scholar
9. Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J: Assembly of human neuronal nicotinic receptor a5 subunits with α3, β2, and β4 subunits. J Biol Chem 1996; 271:17656–17665Google Scholar
10. Sivilotti LG, McNeil DK, Lewis TM, Nassar MA, Schoepfer R, Colquhoun D: Recombinant nicotinic receptors, expressed in Xenopus oocytes, do not resemble native rat sympathetic ganglion receptors in single-channel behaviour. J Physiol 1997; 500(pt 1):123–138 Google Scholar
11. Yu CR, Role LW: Functional contribution of the α5 subunit to neuronal nicotinic channels expressed by chick sympathetic ganglion neurones. J Physiol 1998; 509(pt 3):667–681Google Scholar
12. Gerzanich V, Wang F, Kuryatov A, Lindstrom J: α5 subunit alters desensitization, pharmacology, Ca++ permeability, and Ca++ modulation of human neuronal α3 nicotinic receptors. J Pharmacol Exp Ther 1998; 286:311–320Google Scholar
13. Girod R, Crabtree G, Ernstrom G, Ramirez-Latorre J, McGehee D, Turner J, Role L: Heteromeric complexes of α5 and/or α7 subunits: effects of calcium and potential role in nicotine-induced presynaptic facilitation. Ann N Y Acad Sci 1999; 868:578–590Google Scholar
14. Groot-Kormelink PJ, Boorman JP, Sivilotti LG: Formation of functional α3β4α5 human neuronal nicotinic receptors in Xenopus oocytes: a reporter mutation approach. Br J Pharmacol 2001; 134:789–796 Google Scholar
15. Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR: Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 2004; 65:1526–1535Google Scholar
16. Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C: Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 2002; 22:8785–8789Google Scholar
17. Bierut LJ, Schuckit MA, Hesselbrock V, Reich T: Co-occurring risk factors for alcohol dependence and habitual smoking. Alcohol Res Health 2000; 24:233–241Google Scholar
18. Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, Reich T, Schmidt I, Schuckit MA: A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol 1994; 55:149–158Google Scholar
19. Bierut LJ, Rice JP, Goate A, Hinrichs AL, Saccone NL, Foroud T, Edenberg HJ, Cloninger CR, Begleiter H, Conneally PM, Crowe RR, Hesselbrock V, Li TK, Nurnberger JI Jr, Porjesz B, Schuckit MA, Reich T: A genomic scan for habitual smoking in families of alcoholics: common and specific genetic factors in substance dependence. Am J Med Genet A 2004; 124:19–27Google Scholar
20. Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO: The Fagerström Test for Nicotine Dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict 1991; 86:1119–1127Google Scholar
21. Reich T, Edenberg HJ, Goate A, Williams JT, Rice JP, Van Eerdewegh P, Foroud T, Hesselbrock V, Schuckit MA, Bucholz K, Porjesz B, Li TK, Conneally PM, Nurnberger JI Jr, Tischfield JA, Crowe RR, Cloninger CR, Wu W, Shears S, Carr K, Crose C, Willig C, Begleiter H: Genome-wide search for genes affecting the risk for alcohol dependence. Am J Med Genet 1998; 81:207–215Google Scholar
22. Foroud T, Edenberg HJ, Goate A, Rice J, Flury L, Koller DL, Bierut LJ, Conneally PM, Nurnberger JI, Bucholz KK, Li TK, Hesselbrock V, Crowe R, Schuckit M, Porjesz B, Begleiter H, Reich T: Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping. Alcohol Clin Exp Res 2000; 24:933–945Google Scholar
23. Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, Chen Z, Chu J, Carcassi C, Contu L, Du R, Excoffier L, Ferrara GB, Friedlaender JS, Groot H, Gurwitz D, Jenkins T, Herrera RJ, Huang X, Kidd J, Kidd KK, Langaney A, Lin AA, Mehdi SQ, Parham P, Piazza A, Pistillo MP, Qian Y, Shu Q, Xu J, Zhu S, Weber JL, Greely HT, Feldman MW, Thomas G, Dausset J, Cavalli-Sforza LL: A human genome diversity cell line panel. Science 2002; 296:261–262Google Scholar
24. Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW: Genetic structure of human populations. Science 2002; 298:2381–2385Google Scholar
25. Hinrichs AL, Wang JC, Bufe B, Kwon JM, Budde J, Allen R, Bertelsen S, Evans W, Dick D, Rice J, Foroud T, Nurnberger J, Tischfield JA, Kuperman S, Crowe R, Hesselbrock V, Schuckit M, Almasy L, Porjesz B, Edenberg HJ, Begleiter H, Meyerhof W, Bierut LJ, Goate AM: Functional variant in a bitter-taste receptor (hTAS2R16) influences risk of alcohol dependence. Am J Hum Genet 2006; 78:103–111Google Scholar
26. Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21:263–265Google Scholar
27. Lange C, Silverman EK, Xu X, Weiss ST, Laird NM: A multivariate family-based association test using generalized estimating equations: FBAT-GEE. Biostatistics 2003; 4:195–206Google Scholar
28. Karadsheh MS, Shah MS, Tang X, Macdonald RL, Stitzel JA: Functional characterization of mouse α4β2 nicotinic acetylcholine receptors stably expressed in HEK293T cells. J Neurochem 2004; 91:1138–1150Google Scholar
29. Vernon WI, Printen JA: Assay for intracellular calcium using a codon-optimized aequorin. Biotechniques 2002; 33:730, 732, 734Google Scholar
30. Filatov GN, White MM: The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol 1995; 48:379–384Google Scholar
31. Groot-Kormelink PJ, Luyten WH, Colquhoun D, Sivilotti LG: A reporter mutation approach shows incorporation of the “orphan” subunit β3 into a functional nicotinic receptor. J Biol Chem 1998; 273:15317–15320Google Scholar
32. Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA: Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature 1995; 376:514–516Google Scholar
33. Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP: Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 1991; 353:846–849Google Scholar
34. Marks MJ, Smith KW, Collins AC: Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther 1998; 285:377–386Google Scholar
35. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the folin phenol reagent. J Biol Chem 1951; 193:265–275Google Scholar
36. Kim H, Flanagin BA, Qin C, Macdonald RL, Stitzel JA: The mouse Chrna4 A529T polymorphism alters the ratio of high to low affinity α4β2 nAChRs. Neuropharmacology 2003; 45:345–354 Google Scholar
37. Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V: α-5/ α-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry 2008; 13:368–373Google Scholar
38. Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KK, de Vegt F, Mulders PF, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, Stefansson K: A variant associated with nicotine dependence, lung cancer, and peripheral arterial disease. Nature 2008; 452:638–642Google Scholar
39. Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS: Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet (Epub ahead of print, April 2, 2008)Google Scholar
40. Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P: A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008; 452:633–637Google Scholar
41. Kubota T, Nakajima-Taniguchi C, Fukuda T, Funamoto M, Maeda M, Tange E, Ueki R, Kawashima K, Hara H, Fujio Y, Azuma J: CYP2A6 polymorphisms are associated with nicotine dependence and influence withdrawal symptoms in smoking cessation. Pharmacogenomics J 2006; 6:115–119Google Scholar
42. Audrain-McGovern J, Al Koudsi N, Rodriguez D, Wileyto EP, Shields PG, Tyndale RF: The role of CYP2A6 in the emergence of nicotine dependence in adolescents. Pediatrics 2007; 119:e264–e274Google Scholar
43. Butt CM, King NM, Lauderbaugh AM, Wehner JM, Collins AC: Role of α4, α5, and β2 nicotinic subunits in acetylcholine-stimulated [ 3 H]-aminobutyric acid release from thalamic, striatal, and hippocampal synaptosomes. Washington, DC, Society for Neuroscience, 2005 Abstract Viewer/Itinerary Planner, program no 953.13. http://sfn.scholarone.com/itin2005/index.html Google Scholar
44. Breslau N, Johnson EO, Hiripi E, Kessler R: Nicotine dependence in the United States: prevalence, trends, and smoking persistence. Arch Gen Psychiatry 2001; 58:810–816Google Scholar
45. Kandel DB, Chen K: Extent of smoking and nicotine dependence in the United States: 1991–1993. Nicotine Tob Res 2000; 2:263–274Google Scholar

