
Lower Prepulse Inhibition in Children With the 22q11 Deletion Syndrome
Abstract
OBJECTIVE: The 22q11 deletion syndrome is associated with a range of possible physical anomalies, probable ongoing learning disabilities, and a specific constellation of neuropsychological deficits, including impairments in selective and executive visual attention, working memory, and sensorimotor functioning. It has been estimated that 25% of the children with 22q11 deletion syndrome go on to develop schizophrenia in late adolescence or adulthood. This is of urgent concern. Specification of early brain network vulnerabilities may provide a basis for early intervention while indicating critical links between genes and severe psychiatric illness. Neuropsychological studies of children with 22q11 deletion syndrome have implicated an array of potentially aberrant brain pathways. This study was conducted to determine whether preattentive processing (“sensorimotor gating”) deficits are present in this population. METHOD: The authors administered a test of prepulse inhibition to 25 children with 22q11 deletion syndrome and their 23 sibling comparison subjects, ages 6–13. It was predicted that the children with 22q11 deletion syndrome would have lower prepulse inhibition than the comparison subjects. RESULTS: Prepulse inhibition in the children with 22q11 deletion syndrome (26.06%) was significantly less than that of the sibling comparison subjects (46.41%). Secondary analyses suggested that this decrement did not reflect developmental delay, and lower prepulse inhibition was associated with particular subsyndromal symptoms in some children. CONCLUSIONS: Sensorimotor gating is lower in children with 22q11 deletion syndrome. These findings may indicate specific brain circuits that are anomalous in 22q11 deletion syndrome.
The 22q11 deletion syndrome results from a meiotic deletion of genetic material at the q11.2 site on chromosome 22. It occurs in 1 of every 6,000 births (1) and in over 90% of the cases is de novo (2). Associated congenital anomalies occur in some but not all children and may include heart defects, immunologic deficits, craniofacial dysmorphologies, velopharyngeal defects such as overt or submucous cleft palate, or inflammation-related pain syndromes (3). Before the identification of a single underlying deletion, the different clinical labels described a child’s primary physical anomaly and included “conotruncal anomaly face syndrome” (heart defect with facial dysmorphologies), “velocardiofacial syndrome” (velopharyngeal, heart, and facial anomalies), and “DiGeorge syndrome” (immunologic insufficiency).
Broadly speaking, the academic and neuropsychological profile of children with 22q11 deletion syndrome is more consistent than their physical phenotype. Gross motor, fine motor, and expressive language delays have been identified in the early years (4) and are followed by learning disabilities and academic failures, attention impairment, and behavioral anomalies in an estimated 90%–100% of the school-age children with this syndrome (5, 6). Of greatest concern, retrospective adult studies have suggested that approximately 25% of the children with 22q11 deletion syndrome go on to develop schizophrenia in late adolescence or early adulthood (7, 8). This outcome dramatically increases the significance of their neurocognitive status during the presymptomatic childhood years. Specifying, differentiating, and following the evolution of neurocognitive deficits in children with 22q11 deletion syndrome could eventually provide the basis for linking genetic factors, brain functioning, and severe psychiatric outcome, as well as provide targeted strategies for early intervention.
Neuropsychological studies have hinted at the brain pathways most vulnerable in children with 22q11 deletion syndrome. Marked deficits have been found in visual location memory and working memory (9, 10), selective and executive visual attention (11–13), and sensorimotor functioning (13), perhaps suggesting impairment in the prefrontal cortex and in pathways linking prefrontal and subcortical regions. However, neuropsychological tests alone can lack functional specificity. Psychophysiologic measures, in particular startle inhibition, provide an empirical measure of prefrontal influences on subcortical and brainstem processing.
When a startle-eliciting stimulus is preceded by a relatively low-intensity short-lead stimulus, the strength and the latency of the startle eye blink are inhibited (14). The primary acoustic startle circuit in humans includes the auditory nerve, ventral cochlear nucleus, pontine reticular formation, and spinal motor neurons (15, 16), and it is within this circuit that both startle and startle inhibition originate. The amount of startle inhibition, however, is modulated by descending forebrain circuitry through the limbic cortex, striatum, and pallidum and into the pontine tegmentum and primary perceptual startle circuit (17). This frontal influence on the “automatic” brainstem startle response is believed to protect sensory processing of the lead stimulus (14). Because the startle reflex is a motor response, this phenomenon has been referred to as “sensorimotor gating” (18). Latency of response peak and onset are also modulated by lead stimuli. However, while inhibition occurs reliably among adults at short-lead interval durations ranging from 30 to 500 msec (19), latency effects appear far more sensitive to short-lead interval durations (i.e., 120 versus 250 msec), especially in children (20, 21). The neural pathways that influence latency are elusive, however, and response inhibition (prepulse inhibition) has become better defined.
The evolution of prepulse inhibition in infants and children has been described (20–22) and appears to parallel the developmental stages of brain inhibitory mechanisms (23). Prepulse inhibition in children has been shown to approach adult levels (50%–75%) by approximately age 8, and by this time the gender differences observed in adults are also apparent, with prepulse inhibition values in males 25%–30% higher than those in females (20, 21).
Consistent with the neurodevelopmental findings, lower prepulse inhibition has been shown among children with disorders characterized by a failure of inhibitory brain mechanisms, including Tourette’s syndrome (24), posttraumatic stress disorder (25), fragile X syndrome (26), and, among boys, nocturnal enuresis and comorbid attention deficit hyperactivity disorder (ADHD) (27). Lower prepulse inhibition has been found in adults whose psychiatric syndromes are characterized by poor selective-inhibitory control of attention, including obsessive-compulsive disorder (28, 29), panic disorder (30), social phobia (31), Asperger’s syndrome (32), Huntington’s disease (33), bipolar mania (34), and perhaps most notably, schizophrenia (35). Thus, lower prepulse inhibition appears to occur among individuals who are subject to unfiltered and irrelevant sensory, cognitive, and/or motor perceptions (28, 36). This is especially relevant with regard to schizophrenia. Differences in prepulse inhibition have provided a well-replicated neurophysiologic measure of sensorimotor gating differences in patients with schizophrenia that have been associated with overawareness and misinterpretation of preconscious material (37, 38) and, more generally, the cardinal symptoms of cognitive fragmentation and thought disorder (39, 40).
We explored the possibility of prepulse inhibition differences in children with 22q11 deletion syndrome. We administered a prepulse inhibition procedure to 25 affected children and 23 sibling comparison subjects and predicted that the children with 22q11 deletion syndrome would have significantly lower prepulse inhibition than the sibling comparison subjects. Given the mean age of the subjects (9.2 years), we examined but did not predict latency facilitation effects, which have been shown to be variable during these ages (20, 21). On the basis of past findings (41), we hypothesized gender differences for the sibling comparison subjects, i.e., that the girls would have less prepulse inhibition than the boys.
Method
Participants
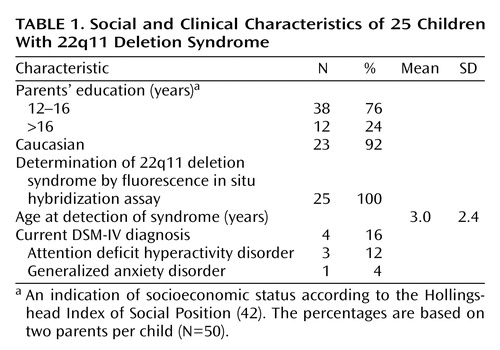
We present data from 48 children between the ages of 6.0 and 13.5 years. One group comprised 25 children with 22q11 deletion syndrome whose deletions were confirmed before study enrollment by means of fluorescence in situ hybridization; the group included 13 boys (mean age=9.1 years, SD=2.3) and 12 girls (mean age=8.7, SD=1.8). The other group included 23 unaffected sibling comparison subjects, nine boys (mean age=10.5, SD=1.9) and 14 girls (mean age=8.3, SD=2.1), who were without a history of learning disability, neuropsychological impairment, and neurologic or psychiatric disorder. All of the girls in the study were prepubescent. The difference in mean age (0.2 year) between the affected and comparison groups was not significant (t=0.38, df=46, p=0.70). Table 1 provides social and clinical characteristics of the affected children.
A total of 55 children attempted the procedure for measuring prepulse inhibition. All of the sibling comparison subjects (N=23) responded to and completed the procedure. However, of the 32 children with 22q11 deletion syndrome, three (two girls, one boy) were not responders (i.e., no blink was detected in more than 33% [six] of the pulse trials); one girl also interrupted the procedure before completion. With regard to hearing ability, all 23 of the sibling comparison subjects and 18 (62%) of the 29 children with 22q11 deletion syndrome were without history of chronic otitis, other transient ear infection, fluid in the ears, ear tubes, or other ear-related difficulties and had had normal results on all hearing tests previously administered. Of the 11 children with 22q11 deletion syndrome who had a history of transient or chronic otitis, eight had no occurrences of infection, fluid in the ears, ear-related problems, or insertion of ear tubes after age 5 and were determined to have had normal hearing ability for 3 or more consecutive years before testing (i.e., the children included in this study group who were under 8 years of age had no history of otitis). Four children with 22q11 deletion syndrome (two boys, two girls) who completed the prepulse inhibition procedure were dropped from analysis because unequivocal proof of normal hearing ability was not available or otitis had not yet completely resolved.
To determine the children’s diagnostic status at the time of testing, their parents completed the Child Symptom Inventory, a diagnostic screening instrument with demonstrated reliability, discriminant validity, and 4-year temporal stability for identifying children with behaviors that may be associated with DSM-IV diagnoses (43). For the children who had positive screening results, the NIMH Diagnostic Interview Schedule for Children, version IV (44), was completed with the parents to ascertain whether the full diagnostic criteria for DSM-IV diagnoses were met. No comparison subjects and four of the 25 children with 22q11 deletion syndrome met criteria for DSM-IV disorders, including ADHD (N=3) and generalized anxiety disorder (N=1) (Table 1).
To demonstrate the comparability of electromyographic (EMG) and photoelectric cell recordings of eye-blink response, seven adult participants of normal hearing ability (four women and three men, 24–40 years of age) performed the prepulse inhibition procedure with simultaneous photoelectric and EMG recordings under the same conditions as those described in the following.
Apparatus
All data were acquired with the Startle Reflex System (series N, model N) from San Diego Instruments (San Diego). Two validated methods were available for detecting the eye-blink startle response (45). The photoelectric cell method records eyelid movement by means of a 10-cm light-emitting diode (LED). Lid closure reflects a LED ray back to an adjacent photocell pickup; millivolt output increases with movement initiation and decreases with blink completion, and a waveform is produced. The EMG method records change at the millivolt level of the orbicularis oculi muscle in response to a startle stimulus by means of surface electrodes attached to the skin around the eye. The startle reflex system was configured to collect both outputs simultaneously.
The photoelectric method requires approximately 2 minutes for fitting the headband apparatus and is noninvasive. The EMG method requires approximately 15 minutes of preparation, including light abrasion of the skin around the eye and behind the ear (with alcohol or abrasive cream), adhesion of electrodes to the eye area and back of the earlobe, and removal of electrodes, reabrasion, and electrode readhesion depending on impedance testing results. Young children (<8 years) in general and most children with 22q11 deletion syndrome can be physically restless, wary, and/or extremely sensitive to touch. Additionally, children with 22q11 deletion syndrome undergo multiple medical screens and procedures once their deletion is detected, leaving them easily frightened by and noncompliant with atypical procedures. To ensure the comfort of the children in this study and high participant compliance, and thus acceptable subject representation, the photoelectric method was selected. The close comparability of the photoelectric and EMG methods for startle modification tests has been previously demonstrated (45). Because the photoelectric method has not been used frequently for prepulse inhibition studies, however, we also tested the comparability of the photoelectric and EMG methods for assessing prepulse inhibition in the procedure here reported.
EMG Data Acquisition
Maximum reduction of electrical impedance is critical for EMG validity and reliability. The skin around the right eye and behind the right ear was abraded with alcohol scrubs for approximately 30 seconds. Three 4-mm silver chloride electrodes were used. Electrode gel was applied to improve conduction. Electrodes were attached to the skin surface with adhesive rings below the lower eyelid (over the orbicularis oculi muscle), at the corner of the same eye, and behind the earlobe. Impedance levels were obtained for each electrode. If the impedance was found to be above 2000 Ω, the electrodes were removed, the skin was reabraded, and the electrodes were replaced. The procedure was repeated as needed (two or three placements per participant were required). When acceptable impedance was obtained, adequate electrode placement was confirmed by monitoring voluntary and involuntary eye blinks on the system’s internal oscilloscope (1–3 minutes). A minimum of 15 minutes was required for electrode placement prior to the initiation of the startle procedure.
Photoelectric Cell Data Acquisition
This system required fitting a padded headband, onto which was affixed (by means of 1-cm wire) a 10-cm rectangular LED/photoelectric cell unit. The unit was positioned parallel to the upper right eyelid at a distance of approximately one-eighth inch and approximately 20° off the eye center. The headband apparatus required approximately 30 seconds to fit and adjust. Positioning of the LED/photoelectric cell unit at the level of the eyelid required an additional 1–2 minutes, during which voluntary and involuntary eye blinks were monitored on the system’s internal oscilloscope.
Auditory Stimuli
Auditory stimuli were presented binaurally through headphones, which were nested over the LED headband. Background white noise of 56 dB (A scale) was present before, between, and after all trial stimulus presentations. Eye blink was elicited with a 50-msec white noise sound burst at 104 dB. The lead interval was fixed at 100 msec (19) for all prepulse trials, with a 70-dB white noise (46) prestimulus followed by a 50-msec white noise sound burst at 104 dB. The software was programmed to deliver one session of 12 minutes’ duration beginning with a 60-second white-noise-only acclimation period, followed by 36 individual stimulus trials, 18 pulse and 18 prepulse trials. The trial types and intertrial intervals, ranging from 12 to 22 seconds, were randomly distributed in a split-half design to ensure equal representation in each half.
Procedure
All prepulse inhibition sessions were completed before 1:00 p.m. to control for circadian effects. The parents and children in the study were first informed of the study tests and procedures 1 month prior to testing by means of mailed consent forms. Just before the start of testing, the prepulse inhibition procedure and apparatus were described and consent was obtained. For the children this occurred with the parents present during the informed consent/child assent procedure. At the time of testing, the participant was seated in a cushioned armchair approximately 4 feet from a 20-inch television monitor and told that he or she would be watching silent cartoons throughout the procedure. The presentation of cartoons during prepulse inhibition has been shown to reduce movement and engage children’s interest (20, 21). For comparability, the same distractor was used with adult participants. The apparatus was handed to the participant to examine, modeled by the experimenter, and then fitted on the participant. The experimenter sat behind the participant, positioned to monitor both the participant and the apparatus throughout the session. For children, two prepulse inhibition sessions were attempted, one per morning for two consecutive mornings of testing. For the adult comparability study, one session per adult was obtained.
Data Analysis
All databases were created, maintained, and analyzed with Statview/PC Version 5 and SAS Version 6.0 (SAS Institute, Cary, N.C.). Variables from the pulse and prepulse trials included the following: millivolt level at the start of the trial, maximum amplitude of the peak in millivolts, time of the peak in milliseconds, inhibition effects calculated as the maximum amplitude of the peak for pulse trials minus the maximum amplitude of the peak for prepulse trials, and the percentage of the amplitude decrease, or prepulse inhibition, calculated as 100 – ([maximum in prepulse trials/maximum in pulse trials] × 100). The latency effect was calculated as the time of the peak in pulse trials minus the time of the peak in prepulse trials, with positive scores reflecting an earlier response peak in the prepulse trials (latency facilitation). Differences in these variables between the photoelectric and EMG methods were examined with paired t tests (p≤0.10). Because the millivolt value ranges for the photoelectric and EMG methods differed, the raw values of millivolt-based variables were transformed to t scores (mean=50, SD=10); such transformation was not necessary for millisecond-based variables. For preliminary analyses of the primary data set, paired t tests were used to examine the comparability of sessions 1 and 2. The F test for equality of variances and the Kolmogorov-Smirnov test (47, 48) were used to examine distribution properties and normality. The one variable with unequal variances in the two groups (time of peak in pulse trials) was successfully normalized by using log10. Group variances of its complement variable (time of peak in prepulse trials) were not unequal, but values for this variable were also log-transformed to allow for the calculation of inhibition effects (as already described). Exponentiated values for logged means are reported. Unpaired t tests were used to assess the comparability of the performance of the children with 22q11 deletion syndrome and the sibling comparison subjects. Analysis of variance (ANOVA) models were used to examine group and gender differences for prepulse inhibition and response latency. Linear regression was used to examine possible age effects on the primary outcome variable. Unpaired t tests were used in secondary analyses to examine differences in prepulse inhibition among children with and without syndromal and subsyndromal symptoms.
Comparability of EMG and Photoelectric Cell Methods
To demonstrate the comparability of recording methods, data were simultaneously collected from the electrode and photoelectric cell recording outputs. Seven adult participants completed one session each. The mean of electrode impedance over seven sessions multiplied by three electrodes was 1.4 (range=0.8–2.4). The variables analyzed for this comparison included the following: millivolt level at the start of the pulse and prepulse trials, maximum amplitude of the peak in millivolts for the pulse and prepulse trials, time of the peak in milliseconds for the pulse and prepulse trials, and percentage prepulse inhibition (Table 2). At p≤0.10, no significant differences between variables from the photoelectric and EMG recording methods were found. The only difference that appeared questionable was that for the time of the peak for the prepulse trials only (12-msec difference). When the raw data were reexamined, it appeared that for sessions where the EMG response was low (5 to 10 mV) the waveform from photoelectric testing indicated a peak of low-voltage eyelid activity between approximately 20 msec and 40 msec after stimulus presentation. This difference did not influence the mean values for prepulse inhibition percentage, which were nearly identical for the two methods. The implications of these findings are further considered in the Discussion section.
Data Evaluation
Prior to data entry all trials for all sessions were individually examined. First trials used the pulse alone and were eliminated to reduce the influence of “first-time” effects. The latency, or response onset, is typically approximated at 30–50 msec (49). Trials with high (>500) or rising millivolt levels within 0–20 msec after stimulus presentation indicated spontaneous eye activity unrelated to startle stimuli and were excluded as “noise” trials. With the noise trials excluded, all sessions were reviewed to determine responder status. “Nonresponder” was defined as a subject who blinked in response to pulses in fewer than 33% of the pulse trials (six of 18), and these sessions were excluded from further analysis. Paired t tests were used to examine the comparability of sessions conducted on consecutive mornings, and no difference on any of the variables analyzed was found. In the majority of cases, the data from day 2 included fewer noise trials, and the tester’s observational reports of child behavior on day 2 reflected notable decreases in wariness and increases in comfort level and behavioral stillness. For these analyses, the data from the day 2 sessions were used (one session per child).
The comparability of the two groups in terms of excluded (“noise”) and included trials and in terms of trial performance was examined. The numbers of pulse and prepulse noise trials did not differ significantly between the subjects with 22q11 deletion syndrome and the sibling comparison subjects: the mean numbers of pulse noise trials were 3.44 (SD=1.87) and 3.39 (SD=2.04), respectively (t=0.09, df=46), and the mean numbers of prepulse noise trials were 2.80 (SD=2.55) and 3.87 (SD=2.70), respectively (t=1.44, df=46). The number of pulse trials that did not elicit a blink response did not differ significantly between groups; however, there was a nearly significant difference in the number of no-response pulse trials between the group with 22q11 deletion syndrome (mean=2.12, SD=2.49) and the comparison subjects (mean=0.96, SD=1.64) (t=1.90, df=46, p=0.07). The numbers of pulse and prepulse trials eliciting a blink response did not differ between the group with 22q11 deletion syndrome and the comparison group; the mean numbers for the pulse trials were 12.44 (SD=3.28) and 13.65 (SD=2.57), respectively (t=1.42, df=46), and the mean numbers for the prepulse trials were 10.84 (SD=4.90) and 10.57 (SD=3.84), respectively (t=0.22, df=46). To further check for potentially confounding performance differences, we compared the two groups’ pulse and prepulse millivolt levels at the start of the trial and the maximum amplitudes of the peaks. No differences were found between the 22q11 deletion syndrome group and the sibling comparison group; the mean millivolt levels at the start of the pulse trials were 190.97 (SD=87.30) and 199.32 (SD=82.40), respectively (t=0.34, df=46), the millivolt levels at the start of the prepulse trials were 192.03 (SD=78.77) and 181.78 (SD=72.16), respectively (t=0.47, df=46), and the mean maximum amplitudes of the peaks in the pulse trials were 3170.35 (SD=1435.42) and 2947.20 (SD=1237.38), respectively (t=0.58, df=46).
Descriptive statistics for key variables are shown in Table 3. The distributions of all variables were examined for bimodality and analyzed for normality. The data for all variables in both groups were found to be normally distributed (Kolmogorov-Smirnov statistics, Table 3). We also performed an F test comparison for equality of variances for each variable. The variances for one of the eight variables examined were found to differ significantly. The children with 22q11 deletion syndrome had markedly greater variance in mean time to peak amplitude in the pulse trials than did the sibling comparison subjects; their variances were 91.44 and 35.05 msec, respectively (variance ratio test, F=0.38, df=22, 24, p=0.03). This variable was transformed to log10, and this adequately equalized the variances (variance ratio test, F=0.66, df=22, 24, p=0.19).
Results
A total of 48 sessions were included in these analyses, 25 from children with 22q11 deletion syndrome and 23 from sibling comparison subjects. The variances in percentage prepulse inhibition were determined to be approximately equivalent between groups (variance ratio test, F=0.77, df=22, 24, p=0.54). An ANOVA was used to examine the primary hypothesis that children with 22q11 deletion syndrome have less prepulse inhibition than sibling comparison subjects. The model (N=44, mean square error=738.02) compared groups by gender. The effect of diagnosis was significant (F=7.83, η=0.39, df=3, 44, p=0.008) and suggested that, overall, the children with 22q11 deletion syndrome had significantly less prepulse inhibition than the comparison subjects. The mean percentages of prepulse inhibition were 26.06% and 46.41%, respectively, and the difference between these values is 20.35%, with a 95% confidence interval of 4.35–36.35 (Fisher’s protected least significant difference test, critical difference=15.82, p=0.02). No independent effect of gender was found in this model (F=0.50, η=0.10, df=3, 44, p=0.48), but a nearly significant interaction of diagnosis and gender was indicated (F=2.81, η=0.23, df=3, 44, p=0.10). On further examination it was found that the group effect was more apparent among boys, with the mean percentage prepulse inhibition for the male comparison subjects within the range of adult levels (57.93%) and the percentage for the boys with 22q11 deletion syndrome significantly less (22.34%). The mean percentages of prepulse inhibition for the comparison girls and girls with 22q11 deletion syndrome differed less (39.00% and 30.09%, respectively) (Figure 1).
The characteristics of the latency difference were examined. The variances of the two groups for this variable did not differ significantly (variance ratio test, F=0.49, df=22, 24, p=0.10). An ANOVA was used to compare the latency difference in the children with 22q11 deletion syndrome and the sibling comparison subjects by gender. There was a significant effect only for gender (F=6.04, df=3, 44, p=0.02) and no effect for diagnosis (F=1.70, df=3, 44, p=0.20) or the interaction of gender and diagnosis (F=0.01, df=3, 44, p=0.94); latency facilitation was apparent in the boys but not in the girls.
An unpaired t test was conducted to examine gender effects among the comparison subjects for percentage prepulse inhibition. Consistent with the results of past studies (20, 21), the difference in the percentage of prepulse inhibition between the unaffected male and female comparison subjects was in the predicted direction and of the expected magnitude. The female comparison subjects exhibited substantially less prepulse inhibition than the male comparison subjects: the mean percentages of prepulse inhibition were 39.00% (SD=26.22) and 57.93% (SD=20.97), respectively, and the difference between the means was 18.93%, but the effect fell short of significance (t=1.82, df=21, p=0.09). Given the magnitude of the difference observed, the lack of significance may be attributable to the small number of subjects and unequal cells (nine boys, 14 girls).
Past studies have suggested that prepulse inhibition increases developmentally until approximately age 8 (20, 21). This study group included children both older and younger than 8 years. If the percentage of prepulse inhibition among the children with 22q11 deletion syndrome increases with age, their lower scores could be attributable to developmental delay rather than consistent impairment. Using linear regression, we attempted to predict the percentage of prepulse inhibition from age in our 25 children with 22q11 deletion syndrome. The slope of the regression line was not significantly greater than zero and did not indicate an association between age and prepulse inhibition in the 22q11 deletion group (slope=–0.243, t=–0.08, df=23, p=0.94). For comparison, we conducted the same analysis in our 23 sibling comparison subjects. The results were not significant, but the association approached significance, with the percentage of prepulse inhibition appearing to increase between ages 6 and 13 among the comparison subjects (slope=4.44, t=1.96, df=21, p=0.07).
Secondary exploratory analyses were used to examine whether prepulse inhibition in the children with 22q11 deletion syndrome was associated with any specific behavioral sign or symptom. We first compared the percentages of prepulse inhibition in children who did and did not meet DSM criteria (Table 1) and found no significant difference (difference between means=7.16%, t=0.48, df=23, p=0.64). We then examined the children’s Child Symptom Inventory severity scores for subsyndromal signs and symptoms in all major categories of childhood disorder. Children can obtain high symptom severity scores but not meet DSM criteria for any disorder. For the more severe disorders (schizophrenia, autism, and Asperger’s syndrome), one or two items result in a high symptom severity score because of the rarity of these symptoms in the normal population. We examined score distributions within subscales and selected for analysis the subscales on which a minimum of five children with 22q11 deletion syndrome scored at or above the 90th percentile (t score of approximately 64 or higher). These included ADHD, generalized anxiety disorder, dysthymia, schizophrenia, autism, and Asperger’s syndrome. We compared the mean percentages of prepulse inhibition for children with higher (≥90%) and lower (≤89%) subscale scores in each of these categories. The percentages for the children with high scores on the subscales for ADHD, generalized anxiety disorder, dysthymia, autism, and Asperger’s syndrome did not differ. However, the mean percentages of prepulse inhibition for children with and without subsyndromal symptoms associated with schizophrenia were 9.04% and 34.07% (difference between means=25.03%, t=2.15, df=23, p=0.05). The symptoms on the Child Symptom Inventory schizophrenia subscale include inappropriate affect, false ideas or beliefs, auditory hallucinations, extremely strange and illogical thoughts or ideas, and extremely odd behavior. Of the 25 children with 22q11 deletion syndrome, eight (32%) had high scores on this scale (five boys, three girls, ages 7–11 years). Six children had one symptom each (either inappropriate affect or extremely odd behavior), and two children had two symptoms (inappropriate affect and illogical beliefs in one child and illogical beliefs and extremely strange ideas in the other child). The possible meaning of this preliminary finding will be considered in the following.
Discussion
Lower prepulse inhibition has been found in child and adult patients whose disorders are characterized by impairment in sensory filtering, selective attention, and inhibitory control. Neuropsychological studies of children with 22q11 deletion syndrome have shown specific impairment in working memory, selective and executive visual attention, and sensorimotor functioning. It has been suggested that 25% of children with 22q11 deletion syndrome go on to develop schizophrenia in early adulthood, and the latter disorder is also characterized by poor selective attention and impaired sensory filtering. To better specify possible sources of early neurocognitive deficits, we assessed prepulse inhibition in children with 22q11 deletion syndrome and sibling comparison subjects.
The children with 22q11 deletion syndrome, as compared with the comparison subjects, had significantly lower prepulse inhibition, suggesting primary sensorimotor gating deficits in the affected children. While a gender effect was not statistically apparent, the difference in the percentage of prepulse inhibition between male subgroups was over three times as large (36%) as that found between female subgroups (9%). The results of a regression analysis of possible age effects were not significant and did not provide evidence that the lower prepulse inhibition among the children with 22q11 deletion syndrome was a function of delay in the development of brain mechanisms associated with prepulse inhibition. Regression analysis among the comparison subjects suggested that prepulse inhibition increased with age, although the effect fell short of significance (p=0.07) and a larger study group is needed to more fully examine developmental effects across this age range and, specifically, to examine the continuing development of prepulse inhibition in females.
Mechanisms
The acoustic startle circuit in humans serially links the auditory nerve, ventral cochlear nucleus, nucleus reticularis pontis caudalis, and spinal motor neurons (50). Prepulse inhibition in rats occurs with prepulse intervals as short as 15 msec; thus, the circuitry responsible for response inhibition cannot involve more than one neuron, or at the most two. It has been suggested that the prepulse activates the ventral cochlear nucleus and nucleus reticularis pontis caudalis. One or both of these activate the pedunculopontine nucleus in the pontine tegmentum, and pedunculopontine nucleus innervation inhibits further response of the nucleus reticularis pontis caudalis (15). The result is reduced startle. While relatively simple, this primary startle/inhibition circuit is actively modulated by a cascade of forebrain structures, including projections from the forebrain limbic cortex into the striatum, striatal projections into the pallidum, and pallidal inputs into the pontine tegmentum (17). The chemoarchitecture associated with these pathways adjusts the level of response inhibition rendered in the pons. Acetylcholine appears critical in the pathways from the medial prefrontal cortex, ventral subiculum, and dentate gyrus, while dopamine, glutamate, and γ-aminobutyric acid (GABA) predominate in projections within and between the caudate nucleus, ventral pallidum, ventral tegmentum, and substantia nigra (36).
Why deficits in prepulse inhibition should occur among children with 22q11 deletion syndrome might be explained in part by the putative neurotransmitter anomalies in these children. The 22q11 deletion syndrome results in the deletion of 30 genes, including two that encode the specific neurotransmitter modulators catechol O-methyltransferase (COMT) and proline dehydrogenase. Studies of COMT inhibitors (51) and knockout mice (52) have demonstrated that COMT fundamentally influences dopamine transmission. Proline dehydrogenase metabolizes proline and also initiates the conversion of proline to glutamate (53, 54). Since glutamate is the precursor of GABA, lowered proline dehydrogenase is likely to produce downstream reduction of GABA levels as well. Neurochemical analysis of mice deficient in proline dehydrogenase supports these predictions, and low prepulse inhibition in these mice substantiates behavioral concomitants of proline dehydrogenase deficiency (55). In addition, l-proline itself seems to function as a modulator of glutamatergic synapses. Thus, dopaminergic, glutamatergic, and GABA-ergic transmission are likely to be dysregulated to various degrees as a result of the loss of COMT and proline dehydrogenase gene alleles in children with 22q11 deletion syndrome. Deletion of additional genes from this region may also contribute to this dysregulation (56 and unpublished 2003 data of J. Mukai et al.), collectively influencing multiple brain pathways, particularly those associated with prepulse inhibition.
Implications
Lower prepulse inhibition may suggest developmental brain pathways that are specifically vulnerable among children with 22q11 deletion syndrome. While a majority (75%) of children with 22q11 deletion syndrome will never develop psychiatric illness, early identification of those who are at highest risk is critical to pursue. For this reason we conducted exploratory comparisons of prepulse inhibition in children with and without current behavioral signs and symptoms. The mean percentages of prepulse inhibition in children with and without diagnosed syndromes did not differ. However, further analyses of children grouped by subsyndromal pathology revealed that children positive for single symptoms associated with schizophrenia had substantially lower prepulse inhibition. The validity of the symptom reports was confirmed with the parents, but this finding requires qualification. Continuity between early subsyndromal symptoms and later outcome has not been suggested in the literature. More important, the specificity of this finding is low. Several children with 22q11 deletion syndrome had low prepulse inhibition and no behavioral symptoms, and one child with a high score on the schizophrenia subscale had high prepulse inhibition. Clearly, lower prepulse inhibition does not alone indicate impairment in individuals or risk of later schizophrenia. Indeed, females in general have lower prepulse inhibition than males. We report this finding only to emphasize the possible significance of subsyndromal signs and to encourage the exploration and meaning of behavioral features that fall outside full-syndromal criteria. Perhaps a combination of lower prepulse inhibition and specific behavioral as well as neurocognitive features will eventually provide our strongest predictor of later risk.
Limitations
Children of all ages tolerated the prepulse inhibition procedure extremely well, and they expressed no discomfort and very little discontent or annoyance with the procedure, despite the nature of the stimuli. While we recognize that the photoelectric cell method has not been frequently used in prepulse inhibition studies, EMG technology for this population was not a reasonable choice of method. Children with the deletion and younger children without the deletion tend to be behaviorally disinhibited, sensitive to touch, and easily frightened, and these characteristics complicate skin preparation, attachment of electrodes to the eye area, and impedance testing. In addition, children with 22q11 deletion syndrome are exposed to multiple early diagnostic and screening procedures for a variety of possible conditions associated with this syndrome. This dramatically increases their later fear and refusal of any procedure that appears foreign, medical, and/or potentially invasive. The motivation for participation in young children is understandably low, and attempting a procedure such as electrode placement can limit or end ongoing compliance of any kind. Child comfort and unbiased subject selection through high compliance were our key priorities.
Our comparability study of seven adults focused only on the variables that would support the validity of the photoelectric cell method for estimating prepulse inhibition. The photoelectric and EMG techniques produced equivalent estimates of prepulse inhibition, indicating that photoelectrically derived prepulse inhibition is in fact comparable to EMG-derived prepulse inhibition. However, these equivalent values for prepulse inhibition were obtained through the measurement of different physiologic phenomena, and additional studies comparing the range of similarities and differences in these methods, and advantages and disadvantages of each, are needed. In fact, the examination of simultaneous recording data suggested interesting new possibilities for the combined use of these methods for testing startle modification. Startle modification procedures provide a powerful method for understanding the developing brain. The photoelectric cell technique provides a child-friendly means of employing this valuable tool with a broad range of child participants and warrants closer examination.
During pilot testing, we found that the use of the silent cartoon distractor was essential for completion of the procedure. Without it, children under the age of approximately 8 would not tolerate the stimuli. In addition, the procedure methods were simplified to the greatest extent possible, and conditions and manipulations that might have provided more information were excluded. For example, while optimal inhibition is achieved at prestimulus intervals between 100 and 150 msec (19), intervals between 15 and 400 msec yield reliable inhibition effects in comparison subjects (57). Among affected participants, variations in response to intervals of different durations may reveal important pathway anomalies, and prepulse inhibition studies often have examined responses under two or more short-lead interval conditions. For this study, however, only one prepulse lead interval was used (100 msec) to ensure a brief procedure (12 minutes) that would be readily tolerated by all children in this study (i.e., for balanced designs, the addition of one interval doubles total session time).
Past studies of prepulse inhibition in children (21) have controlled for IQ. However, cognitive and neuropsychological testing of this study group revealed extremely broad subtest score scatter for each child tested on both IQ and neuropsychological batteries, invalidating the calculation and use of composite IQ scores for this group (13). Selection of a single subtest score would have been arbitrary, and we did not attempt to conceptualize an IQ estimation for these subjects.
The demographic representation in this study group was limited; 90% of the children were Caucasian and of upper-middle or upper social strata. The applicability of these findings to children of other races and/or socioeconomic strata is not known.
 |
 |
 |
Received Oct. 30, 2003; revisions received March 8 and March 29, 2004; accepted May 10, 2004. From the Laboratory of Human Neurogenetics, Rockefeller University. Address correspondence and reprint requests to Dr. Sobin, Rockefeller University, Box 313, 1230 York Ave., New York, NY 10021; [email protected] (e-mail). Supported by grant K08-HD-040321 from the National Institute on Child Health and Human Development (Dr. Sobin) and by General Clinical Research Center grant M01-RR-00102 from the NIH National Center for Research Resources. The authors thank the children in this study for their participation and feedback, the parents for their commitment to their children and to our work, Rosemary Collier for her help in the early stages of data organization and management, Charlie Bubb for his technological guidance, Maude Blundell, M.S., for her contributions to the early recruitment phase of this study, and Kawame Anyane-Yeboa, M.D., for his participant referrals.

Figure 1. Prepulse Inhibition in Boys and Girls With 22q11 Deletion Syndrome and Sibling Comparison Subjects, Based on Eye Blinks Following Startling Auditory Pulses Presented Alone and After Prepulses
1. Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM: A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003; 112(1, part 1):101–107Google Scholar
2. Morrow B, Goldberg R, Carlson C, Das Gupta R, Sirotkin H, Collins J, Dunham I, O’Donnell H, Scambler P, Shprintzen R, Kucherlapati R: Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet 1995; 56:1391–1403Medline, Google Scholar
3. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ, et al: Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 1997; 34:798–804Crossref, Medline, Google Scholar
4. Gerdes M, Solot C, Wang PP, Moss E, LaRossa D, Randall P, Goldmuntz E, Clark BJ III, Driscoll DA, Jawad A, Emanuel BS, McDonald-McGinn DM, Batshaw ML, Zackai EH: Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am J Med Genet 1999; 85:127–133Crossref, Medline, Google Scholar
5. Lipson AH, Yuille D, Angel M, Thompson PG, Vandervoord JG, Beckenham EJ: Velocardiofacial (Shprintzen) syndrome: an important syndrome for the dysmorphologist to recognise. J Med Genet 1991; 28:596–604Crossref, Medline, Google Scholar
6. Shprintzen RJ, Goldberg RB, Young D, Wolford L: The velo-cardio-facial syndrome: a clinical and genetic analysis. Pediatrics 1981; 67:167–172Medline, Google Scholar
7. Murphy KC, Jones LA, Owen MJ: High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry 1999; 56:940–945Crossref, Medline, Google Scholar
8. Pulver AE, Nestadt G, Goldberg R, Shprintzen RJ, Lamacz M, Wolyniec PS, Morrow B, Karayiorgou M, Antonarakis SE, Housman D, et al: Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis 1994; 182:476–478Crossref, Medline, Google Scholar
9. Wang PP, Woodin MF, Kreps-Falk R, Moss EM: Research on behavioral phenotypes: velocardiofacial syndrome (deletion 22q11.2). Dev Med Child Neurol 2000; 42:422–427Crossref, Medline, Google Scholar
10. Bearden CE, Woodin MF, Wang PP, Moss E, McDonald-McGinn D, Zackai E, Emannuel B, Cannon TD: The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J Clin Exp Neuropsychol 2001; 23:447–464Crossref, Medline, Google Scholar
11. Woodin M, Wang PP, Aleman D, McDonald-McGinn D, Zackai E, Moss E: Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet Med 2001; 3:34–39Crossref, Medline, Google Scholar
12. Sobin C, Kiley-Brabeck K, Daniels S, Blundell M, Anyane-Yeboa K, Karayiorgou M: Networks of attention in children with the 22q11 deletion syndrome. Dev Neuropsychol 2004; 26:611–626Crossref, Medline, Google Scholar
13. Sobin C, Kiley-Brabeck K, Daniels S, Khuri J, Taylor L, Blundell M, Anyane-Yeboa K, Karayiorgou M: Neuropsychological characteristics of children with the 22q11 deletion syndrome: a descriptive analysis. Child Neuropsychology 2005; 11:39–53Crossref, Medline, Google Scholar
14. Graham FK: Presidential address, 1974: the more or less startling effects of weak prestimulation. Psychophysiology 1975; 12:238–248Crossref, Medline, Google Scholar
15. Koch M, Lingenhohl K, Pilz PK: Loss of the acoustic startle response following neurotoxic lesions of the caudal pontine reticular formation: possible role of giant neurons. Neuroscience 1992; 49:617–625Crossref, Medline, Google Scholar
16. Koch M: The neurobiology of startle. Prog Neurobiol 1999; 59:107–128Crossref, Medline, Google Scholar
17. Swerdlow NR, Geyer MA: Neurophysiology and neuropharmacology of short lead interval startle modification, in Startle Modification. Edited by Dawson ME, Schell AM, Bohmelt AH. New York, Cambridge University Press, 1999, pp 114–133Google Scholar
18. Braff DL, Geyer MA: Sensorimotor gating and schizophrenia: human and animal model studies. Arch Gen Psychiatry 1990; 47:181–188Crossref, Medline, Google Scholar
19. Graham FK, Putnam LE, Leavitt LA: Lead-stimulation effects of human cardiac orienting and blink reflexes. J Exp Psychol Hum Percept Perform 1975; 104:175–182Medline, Google Scholar
20. Ornitz EM, Guthrie D, Kaplan AR, Lane SJ, Norman RJ: Maturation of startle modulation. Psychophysiology 1986; 23:624–634Crossref, Medline, Google Scholar
21. Ornitz EM, Guthrie D, Sadeghpour M, Sugiyama T: Maturation of prestimulation-induced startle modulation in girls. Psychophysiology 1991; 28:11–20Crossref, Medline, Google Scholar
22. Graham FK, Strock BD, Zeigler BL: Excitatory and inhibitory influences on reflex responsiveness, in Aspects of the Development of Competence: The Minnesota Symposium on Child Psychology. Edited by Collins WA. Hillsdale, NJ, Lawrence Erlbaum Associates, 1981, pp 1–38Google Scholar
23. Ornitz EM: Developmental aspects of neurophysiology, in Child and Adolescent Psychiatry: A Comprehensive Textbook, 3rd ed. Edited by Lewis M. Baltimore, Williams & Wilkins, 2002, pp 87–92Google Scholar
24. Castellanos FX, Fine EJ, Kaysen D, Marsh WL, Rapoport JL, Hallett M: Sensorimotor gating in boys with Tourette’s syndrome and ADHD: preliminary results. Biol Psychiatry 1996; 39:33–41Crossref, Medline, Google Scholar
25. Ornitz EM, Pynoos RS: Startle modulation in children with posttraumatic stress disorder. Am J Psychiatry 1989; 146:866–870Link, Google Scholar
26. Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ: Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 2004; 9:417–425Crossref, Medline, Google Scholar
27. Ornitz EM, Hanna GL, de Traversay J: Prestimulation-induced startle modulation in attention-deficit hyperactivity disorder and nocturnal enuresis. Psychophysiology 1992; 29:437–451Crossref, Medline, Google Scholar
28. Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL: A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biol Psychiatry 1993; 33:298–301Crossref, Medline, Google Scholar
29. Schall U, Schon A, Zerbin D, Eggers C, Oades RD: Event-related potentials during an auditory discrimination with prepulse inhibition in patients with schizophrenia, obsessive-compulsive disorder and healthy subjects. Int J Neurosci 1996; 84:15–33Crossref, Medline, Google Scholar
30. Ludewig S, Ludewig K, Geyer MA, Hell D, Vollenweider FX: Prepulse inhibition deficits in patients with panic disorder. Depress Anxiety 2002; 15:55–60Crossref, Medline, Google Scholar
31. Larsen DK, Norton GR, Walker JR, Stein M: Analysis of startle responses in patients with panic disorder and social phobia. Cogn Behav Ther 2002; 31:156–169Crossref, Google Scholar
32. McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, Schmitz N, Happe F, Howlin P, Murphy DG: Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain 2002; 125(part 7):1594–1606Google Scholar
33. Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR: Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1995; 58:192–200Crossref, Medline, Google Scholar
34. Perry W, Minassian A, Feifel D, Braff DL: Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry 2001; 50:418–424Crossref, Medline, Google Scholar
35. Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR: Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res 2001; 49:171–178Crossref, Medline, Google Scholar
36. Swerdlow NR, Caine SB, Braff DL, Geyer MA: The neural substrates of sensorimotor gating of the startle reflex: a review of recent findings and their implications. J Psychopharmacol 1992; 6:176–190Crossref, Medline, Google Scholar
37. Frith CD: Consciousness, information processing and schizophrenia. Br J Psychiatry 1979; 134:225–235Crossref, Medline, Google Scholar
38. Butler RW, Braff DL: Delusions: a review and integration. Schizophr Bull 1991; 17:633–647Crossref, Medline, Google Scholar
39. McGhie A, Chapman J: Disorders of attention and perception in early schizophrenia. Br J Med Psychol 1961; 34:103–116Crossref, Medline, Google Scholar
40. Braff DL, Grillon C, Geyer MA: Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry 1992; 49:206–215Crossref, Medline, Google Scholar
41. Ornitz EM: Startle modification in children and developmental effects, in Startle Modification. Edited by Dawson ME, Schell AM, Bohmelt AH. New York, Cambridge University Press, 1999, pp 245–266Google Scholar
42. Hollingshead AB, Redlich FC: Social Class and Mental Illness. New York, John Wiley & Sons, 1958Google Scholar
43. Sprafkin J, Gadow KD, Salisbury H, Schneider J, Loney J: Further evidence of reliability and validity of the Child Symptom Inventory—4: parent checklist in clinically referred boys. J Clin Child Adolesc Psychol 2002; 31:513–524Crossref, Medline, Google Scholar
44. Shaffer D, Fisher P, Lucas CP, Dulcan MK, Schwab-Stone ME: NIMH Diagnostic Interview Schedule for Children Version IV (NIMH DISC-IV): description, differences from previous versions, and reliability of some common diagnoses. J Am Acad Child Adolesc Psychiatry 2000; 39:28–38Crossref, Medline, Google Scholar
45. Flaten MA: A comparison of electromyographic and photoelectric techniques in the study of classical eyeblink conditioning and startle reflex modification. J Psychophysiol 1993; 7:230–237Google Scholar
46. Blumenthal TD: Inhibition of the human startle response is affected by both prepulse intensity and eliciting stimulus intensity. Biol Psychol 1996; 44:85–104Crossref, Medline, Google Scholar
47. Kolmogorov AN: Sulla determinazione empirical di una legge di distribuizione. Giornale dell’ Institute Italiano degli Altuari 1933; 4:83–91Google Scholar
48. Daniel WW: Biostatistics: A Foundation for Analysis in the Health Sciences. New York, John Wiley & Sons, 1991, pp 596–602Google Scholar
49. Berg WK, Balaban MT: Methods of startle elicitation, in Startle Modification. Edited by Dawson ME, Schell AM, Bohmelt AH. New York, Cambridge University Press, 1999, pp 21–50Google Scholar
50. Koch M, Kungel M, Herbert H: Cholinergic neurons in the pedunculopontine tegmental nucleus are involved in the mediation of prepulse inhibition of the acoustic startle response in the rat. Exp Brain Res 1993; 97:71–82Crossref, Medline, Google Scholar
51. Napolitano A, Cesura AM, Da Prada M: The role of monoamine oxidase and catechol O-methyltransferase in dopaminergic neurotransmission. J Neural Transm Suppl 1995; 45:35–45Medline, Google Scholar
52. Gogos JA, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, Karayiorgou M: Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci USA 1998; 95:9991–9996Crossref, Medline, Google Scholar
53. Johnson JL, Roberts E: Proline, glutamate and glutamine metabolism in mouse brain synaptosomes. Brain Res 1984; 323:247–256Crossref, Medline, Google Scholar
54. Cohen SM, Nadler JV: Proline-induced inhibition of glutamate release in hippocampal area CA1. Brain Res 1997; 769:333–339Crossref, Medline, Google Scholar
55. Gogos JA, Santha M, Takacs Z, Beck KD, Luine V, Lucas LR, Nadler JV, Karayiorgou M: The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nat Genet 1999; 21:434–439Crossref, Medline, Google Scholar
56. Liu H, Abecasis GR, Heath SC, Knowles A, Demars S, Chen YJ, Roos JL, Rapoport JL, Gogos JA, Karayiorgou M: Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci USA 2002; 99:16859–16864Crossref, Medline, Google Scholar
57. Anthony BJ: In the blink of an eye: implications of reflex modification for information processing, in Advances in Psychophysiology, vol 1. Edited by Ackles PK, Jennings JR, Coles MGH. Greenwich, Conn, JAI Press, 1985, pp 167–218Google Scholar

