
Genome-Wide Association Study of Multiplex Schizophrenia Pedigrees
Abstract
Objective
The authors used a genome-wide association study (GWAS) of multiply affected families to investigate the association of schizophrenia to common single-nucleotide polymorphisms (SNPs) and rare copy number variants (CNVs).
Method
The family sample included 2,461 individuals from 631 pedigrees (581 in the primary European-ancestry analyses). Association was tested for single SNPs and genetic pathways. Polygenic scores based on family study results were used to predict case-control status in the Schizophrenia Psychiatric GWAS Consortium (PGC) data set, and consistency of direction of effect with the family study was determined for top SNPs in the PGC GWAS analysis. Within-family segregation was examined for schizophrenia-associated rare CNVs.
Results
No genome-wide significant associations were observed for single SNPs or for pathways. PGC case and control subjects had significantly different genome-wide polygenic scores (computed by weighting their genotypes by log-odds ratios from the family study) (best p=10−17, explaining 0.4% of the variance). Family study and PGC analyses had consistent directions for 37 of the 58 independent best PGC SNPs (p=0.024). The overall frequency of CNVs in regions with reported associations with schizophrenia (chromosomes 1q21.1, 15q13.3, 16p11.2, and 22q11.2 and the neurexin-1 gene [NRXN1]) was similar to previous case-control studies. NRXN1 deletions and 16p11.2 duplications (both of which were transmitted from parents) and 22q11.2 deletions (de novo in four cases) did not segregate with schizophrenia in families.
Conclusions
Many common SNPs are likely to contribute to schizophrenia risk, with substantial overlap in genetic risk factors between multiply affected families and cases in large case-control studies. Our findings are consistent with a role for specific CNVs in disease pathogenesis, but the partial segregation of some CNVs with schizophrenia suggests that researchers should exercise caution in using them for predictive genetic testing until their effects in diverse populations have been fully studied.
We report here on the first genome-wide association study (GWAS) in families with multiple members with schizophrenia. Significant associations of single-nucleotide polymorphisms (SNPs) can suggest new disease susceptibility mechanisms. For schizophrenia, large GWAS analyses of common SNPs have found associations in the major histocompatibility complex (MHC, chromosome 6) (1–3) and several specific genes (3–5). The Psychiatric GWAS Consortium (PGC) analyzed 21,856 individuals from 17 GWAS samples and then added data from an additional 29,839 individuals (including the present data set) for the most promising findings. The results strongly supported association in seven genes or regions between genes, including the MHC (6). The present study was designed before the typical effect sizes of common SNPs on disease risks became clear (e.g., odds ratios of only 1.1–1.2, conferring a 10%–20% increase in risk), and our sample is now known to be underpowered. However, we can address whether SNPs with larger effects might be “enriched” in families with multiple cases.
The PGC analysis (6) also confirmed a previous finding (1) that is interpreted as suggesting a polygenic effect of many common SNPs on schizophrenia susceptibility, based on the ability of association test results for many SNPs in one data set to predict case versus control status in a second data set. In the present study, we evaluated whether common risk SNPs in multiply affected families are likely to overlap with those in unrelated cases by testing whether our family study results can predict case-control status in the large PGC data set. We also explored whether any known functional gene pathways are enriched for modestly significant SNP associations. In single-SNP, polygenic, and pathway analyses, family data provide some protection against spurious associations due to case-control differences in ancestral backgrounds, because counts of SNP alleles that are transmitted from parents to ill offspring are contrasted with counts of the alleles that parents did not transmit.
GWAS analyses have also shown that rare chromosomal deletions of chromosomes 1q21.1, 15q13.3, and 22q11.2 and of exons of the neurexin-1 gene (NRXN1) and duplications of 16p11.2 (collectively present in around 1.25% of cases) each produce significant eightfold or greater increases in risk; notably, each has also been reported in autism, mental retardation, and epilepsy (7). We determined the frequency of these copy number variants (CNVs) in our families and examined how well they correlate (segregate) with disease in families, which has implications for diagnostic testing. We also identified new “candidate” CNVs.
Method
Subjects
The sample (Table 1) includes seven subsamples that were recruited for linkage studies (8–15) and subsequently combined (16–19), excluding families from the National Institute of Mental Health’s Schizophrenia Genetics Initiative because a previous GWAS studied the probands (2). Briefly, family members gave informed consent and were diagnosed using semistructured interviews, psychiatric records, and informant reports. Case subjects had DSM-III-R diagnoses of schizophrenia or schizoaffective disorder (probands had schizophrenia), which cosegregate in families (20) and are difficult to differentiate reliably (21). These families were originally ascertained because the constellation of affected relatives was informative for linkage studies, and all families had at least two directly evaluated narrow-diagnosis cases. For some families, only one affected case subject was included in this analysis, either because there was only one case subject in the nuclear family who met inclusion criteria or because DNA was not available for GWAS genotyping or the specimen failed quality control filters. Families were analyzed here if they had DNA available for one affected offspring plus one or both parents, for two affected siblings and at least one parent or one unaffected sibling, or for three or more affected siblings. Some families included more than one sibship that met these criteria. Based on an analysis of power versus cost (not shown), we included all available parents plus two unaffected siblings (if available) if no parents were genotyped, or one unaffected sibling if one parent was genotyped.
| Affected and Unaffected Subjects and Ancestry | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotyped Affected | Genotyped Unaffected | Families | ||||||||||||||
| European | Other | European | Other | Other | ||||||||||||
| Site | N | Male | N | Male | N | Male | N | Male | Total | European | Mediterranean | Admix1 | Admix2 | Admix3 | Malabar | Total |
| Australia/United States | 128 | 0.63 | 2 | 0.50 | 107 | 0.49 | 1 | 0.00 | 238 | 57 | 1 | 0 | 0 | 0 | 0 | 58 |
| Cardiff | 43 | 0.70 | 2 | 0.50 | 30 | 0.33 | 2 | 0.50 | 77 | 21 | 0 | 0 | 0 | 0 | 0 | 21 |
| Johns Hopkins | 282 | 0.64 | 6 | 0.67 | 261 | 0.44 | 3 | 0.33 | 552 | 122 | 1 | 0 | 0 | 0 | 0 | 123 |
| Illinois/NorthShore | 118 | 0.74 | 2 | 1.00 | 92 | 0.48 | 1 | 0.00 | 213 | 53 | 1 | 0 | 0 | 0 | 0 | 54 |
| Paris | 53 | 0.57 | 99 | 0.62 | 37 | 0.54 | 83 | 0.36 | 272 | 21 | 0 | 11 | 11 | 5 | 8 | 56 |
| VCU/Ireland | 399 | 0.67 | 0 | — | 286 | 0.43 | 0 | — | 685 | 216 | 0 | 0 | 0 | 0 | 0 | 216 |
| Western Australia/Germany | 195 | 0.52 | 28 | 0.57 | 177 | 0.47 | 24 | 0.50 | 424 | 91 | 12 | 0 | 0 | 0 | 0 | 103 |
| Total | 1,218 | 0.64 | 139 | 0.61 | 990 | 0.45 | 114 | 0.39 | 2,461 | 581 | 15 | 11 | 11 | 5 | 8 | 631 |
Genotyping, SNP Quality Control, and Genotypic Ancestry
Genotyping was performed with the Illumina 610-Quad array (at Illumina, Inc., La Jolla, Calif., for families and at the Children’s Hospital of Philadelphia [by H.H.] for control subjects; see p. 18 of the online data supplement for discussion of the CNV case-control analysis), and genotypes were called with the BeadStudio software package (Illumina, Inc.). HG18 genomic locations are reported. Based on principal components analysis (22) of 55,010 autosomal SNPs with low pairwise linkage disequilibrium (LD), families were divided into six ancestry groups (Table 1; see also Figure S1 in the data supplement that accompanies the online edition of this article): European, Mediterranean (primarily Sephardic Jewish), and four with varying degrees of African or South Indian admixture (Réunion Island). Because somewhat different genetic architecture has been observed for schizophrenia in European- and African-origin samples in previous single-SNP (2) and polygenic (1) GWAS results, separate analyses were carried out for the European-ancestry group and for the six ancestry groups combined.
Exclusion criteria for SNPs were as follows: third allele observed; pseudo-autosomal or mitochondrial; minor allele frequency <1% (in European-ancestry group or all founders); call rate <98.8%; p<0.0001 for deviation from Hardy-Weinberg expectation (in unrelated unaffected individuals); GenCall10 quality score <0.55; and more than four Mendelian inconsistencies for parent-child pairs and more than seven for parent-parent-child trios. Genotypes were removed for the family for SNPs with Mendelian inconsistencies and for males for chromosome X SNPs called as heterozygous. There were 576,976 autosomal and 15,146 chromosome X SNPs before quality control analysis (QC), and 531,195/12,936 for European-ancestry and 528,297/13,202 for all analyses after QC.
DNA sample exclusion criteria were as follows: duplicates of another sample; genotypically inconsistent with known gender or family structure; >104 parent-child or >199 parent-parent-child Mendelian inconsistencies: call rate <98%; or mean heterozygosity inconsistent with ancestry subgroup. Chromosome X data were excluded if genotypic gender was ambiguous (possible cell culture artifact) but autosomal QC was acceptable.
Statistical Analyses of Genetic Association to SNPs
Family-based association tests were performed using TRANSMIT, version 2.5.4 (23), for autosomal SNPs. TRANSMIT was selected because it is fast and can handle any constellation of genotyped relatives. However, it is not recommended for chromosome X, so UNPHASED, version 3.1.5 (24), modified for consistency with TRANSMIT in handling ungenotyped individuals, was used for that chromosome. These programs test whether each SNP allele is transmitted more or less often than chance expectation. Because they use data set allele frequencies as well as the family’s data to estimate nontransmitted alleles of ungenotyped parents, analyses were performed separately for each of the six ancestry subgroups. European-ancestry and all-family results are reported (with the latter combining observed and expected transmission counts across groups). Autosomal odds ratios were estimated by subtracting an estimate of the number of homozygous parents (allele frequency squared, times the number of parents) from the total number of transmissions of each allele to obtain transmissions from heterozygous parents (expected to be 50% for each allele by chance), and computing the ratio of counts for the two alleles. Genomic control lambda was computed as the median chi-square value divided by the expected value (0.456).
Two previous studies noted that TRANSMIT can sometimes inflate type I error (25, 26). One of the studies (26) is difficult to generalize because it used TRANSMIT’s bootstrapping routine to compute p values, which can produce discrete distributions in small samples (37 pedigrees in that study). For the robust variance estimator used here to compute p values, Martin et al. (25) previously clarified that the problem was seen in larger samples when only two affected siblings could be genotyped, in the presence of linkage, and for recessive inheritance with much larger effect sizes than are observed in any GWAS of schizophrenia. We excluded sibling-pair-only families. Also, we initially evaluated TRANSMIT’s type I error rate in 5,000 replicates of our European-ancestry pedigrees for each of a range of minor allele frequencies and linkage models (up to a value of 2 for the relative risk to siblings versus population risk, much stronger than is realistic for schizophrenia) and observed no inflation of type I error rate at nominal significance levels of 0.05–0.001. Finally, our quantile-quantile plots (see Figure S2 in the online data supplement) demonstrate that no substantial inflation occurred.
To estimate power, genotypes were simulated for European-ancestry families under a range of genetic models, and each replicate was analyzed with TRANSMIT. The sample was well powered (>80%) to detect genome-wide significant association for additive allelic relative risks of approximately 1.5 (25%–50% allele frequencies), but not in the range of 1.1–1.2 (1%–2% power to detect genome-wide significant effects).
We performed ALIGATOR (27) analyses of whether gene pathways contained SNPs with low p values more often than would be expected by chance given the observed distribution of SNP p values, for the GO, KEGG, MGI, PANTHER, BioCarta, and Reactome databases plus two locally curated pathways (see p. 12 in the online data supplement).
We used polygenic score tests (1) to evaluate the hypothesis of multiple common risk SNPs, using 112,869 post-QC autosomal SNPs with limited pairwise LD (r2<0.25) that were also available for the PGC phase 1 European-ancestry data set of 9,394 cases and 12,462 controls (using data that were either genotyped or imputed [28] based on HapMap 3 reference haplotypes with information content >0.9). A reference allele for each SNP was assigned a weight equal to the log-odds ratio for association in the family study. For each PGC subject, the observed reference alleles were weighted and summed. The significance of the PGC case-control score difference was analyzed by logistic regression (using the R package), corrected for seven ancestry-based principal component scores as covariates. The proportion of variance explained (R2) by the polygenic scores was computed by subtracting the Nagelkerke R2 attributable to ancestry covariates alone from the R2 for polygenic scores plus covariates. The analysis was repeated 10 times, starting with only the SNPs with the best 0.01% of p values in the family data, and finally including all SNPs (see Figure 2 legend for details).
Finally, the 58 independent (r2<0.2) SNPs with the best p values in the phase 1 PGC GWAS (which did not include the present families) were selected for analysis of consistency of direction of effect in the family study (6). These were drawn from the 81 SNPs with p<2×10−5, including only the best SNP from the extended MHC region that contained most of the significant SNPs but is characterized by extensive LD. For SNPs not genotyped here, we selected a nearby proxy (highest r2 with the PGC SNP). After inverting the family study odds ratios when necessary because of differences in chromosomal strand and/or test allele, we determined the number of SNPs with the same direction (both odds ratios <1 or both >1) in the two analyses and computed a binomial test of the probability of observing at least that many consistencies, given the chance expectation of 50% consistency of direction of effect.
CNV Analysis
Data are presented here for segregation of previously identified schizophrenia-associated CNVs within families (chromosomes 1q21.1, 15q13.3, 16p11.2, and 22q11.2 and NRXN1) (7, 29–31). An exploratory case-control analysis to identify new candidate CNVs was also carried out (for the methods and results, see p. 18 of the online data supplement). CNVs spanning three or more probes were called with the PennCNV software program (32). Subjects were excluded if they had ≥50 CNV calls or if the standard deviation of the log(R) ratio (a normalized expression of relative probe intensity for a given subject, which is related to copy number) was >0.4 (indicating increased signal variability across all probes). CNVs were merged if two or more adjacent deletions or duplications had different estimated copy numbers (0 and 1 for deletions, 3 and 4 for duplications) or if a segment with an estimated copy number of 2 contained <30% of the probes in a CNV formed by merging it with two surrounding deletions or duplications (and these merger rules were also applied to chains of such events). For subjects with one of the schizophrenia-associated CNVs and for all of their family members, CNV data for that region were visualized by plotting log(R) ratio and B-allele frequency (the proportion of intensity detected for a designated test allele) and by computing and visualizing point-by-point estimates of copy number using a second algorithm (33). In all cases, the PennCNV call for these large CNVs was confirmed by these additional steps. For the five selected CNV regions, we then examined evidence for transmission within families and for segregation with schizophrenia.
Results
Association of Common SNPs
For European-ancestry families (Figure 1), lambda (the median chi-square divided by the expected median in null data, 0.456) was 1.025 (see Figure S2 in the online data supplement), indicating minimal technical or ancestry-related artifact. Table 2 lists results for genes with at least one SNP with p<0.0001 within the gene or within 50 kb of it. (See Table S1 in the online data supplement for details of nongenic regions meeting this criterion.) The all-family analysis produced similar results (see Figures S2 and S3 and Table S2 in the online data supplement). No SNP achieved genome-wide significance (p<5×10−8) in either analysis.
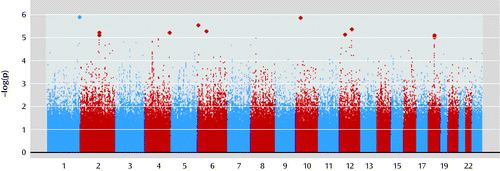
a Each dot represents the −log(p value) for one of the 544,131 autosomal and X chromosome SNPs included in the European-ancestry analysis. Chromosome numbers are shown on the x-axis.
| Allele 1 Total Counts | Allele 2 Total Counts | T From Heterozygous Parents | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | LOC | A1 | Frq | T | NT | A2 | Frq | T | NT | A1 | A2 | Odds Ratio | p | SNPs | Genes (Within 50 kb) |
| rs12210050 | chr6:420489 | T | 0.23 | 565 | 459 | C | 0.77 | 1763 | 1869 | 445.1 | 371.7 | 1.20 | 2.9E-06 | 1 | EXOC2,9648 |
| rs12426725 | chr12:80367259 | A | 0.15 | 308 | 411 | G | 0.85 | 2030 | 1927 | 254.1 | 347.9 | 0.73 | 4.2E-06 | 4 | PPFIA2 |
| rs1170612 | chr2:124699526 | T | 0.22 | 593 | 481 | C | 0.78 | 1745 | 1857 | 475.1 | 339.3 | 1.40 | 5.9E-06 | 7 | CNTNAP5 |
| rs16934812 | chr12:29763585 | G | 0.13 | 331 | 248 | T | 0.87 | 2003 | 2086 | 294.0 | 219.6 | 1.34 | 7.2E-06 | 1 | TMTC1 |
| rs12511372 | chr4:45811189 | G | 0.50 | 1229 | 1106 | A | 0.50 | 1107 | 1230 | 643.8 | 524.2 | 1.23 | 1.4E-05 | 11 | GABRG1 (and GABRA2 in the SNP cluster) |
| rs3197999 | chr3:49696536 | T | 0.30 | 747 | 628 | C | 0.70 | 1587 | 1706 | 539.6 | 437.1 | 1.23 | 1.6E-05 | 1 | BSN,12550; APEH,598; MST1; RNF123,–5457; AMIGO3,33432; GMPPB,37399; IHPK1,40195 |
| rs4716801 | chr7:157381124 | G | 0.46 | 1151 | 1022 | A | 0.54 | 1185 | 1314 | 652.2 | 509.0 | 1.28 | 2.1E-05 | 1 | PTPRN2 |
| rs7805806 | chr7:20693853 | G | 0.12 | 334 | 250 | A | 0.88 | 2004 | 2088 | 298.9 | 203.8 | 1.47 | 2.2E-05 | 1 | ABCB5 |
| rs12239401 | chr1:235261146 | T | 0.44 | 953 | 1080 | C | 0.56 | 1383 | 1256 | 495.2 | 657.5 | 0.75 | 2.2E-05 | 1 | RYR2,–11178 |
| rs6433323 | chr2:172581306 | G | 0.38 | 963 | 842 | A | 0.62 | 1375 | 1496 | 623.6 | 479.2 | 1.30 | 2.3E-05 | 2 | HAT1,24460; MAP1D |
| rs1037231 | chr3:85845797 | A | 0.42 | 913 | 1038 | G | 0.58 | 1421 | 1296 | 506.2 | 629.0 | 0.80 | 3.3E-05 | 4 | CADM2,–12524 |
| rs3892156 | chr16:48877496 | A | 0.25 | 659 | 552 | G | 0.75 | 1669 | 1776 | 509.5 | 371.3 | 1.37 | 3.7E-05 | 1 | ADCY7,–1827; BRD7,32945 |
| rs2396465 | chr2:228234344 | G | 0.10 | 267 | 198 | A | 0.90 | 2071 | 2140 | 244.6 | 168.1 | 1.46 | 3.8E-05 | 1 | DKFZp547H025,–28212; SLC19A3,23825 |
| rs12565770 | chr1:19427647 | A | 0.12 | 229 | 312 | G | 0.88 | 2109 | 2026 | 197.6 | 281.8 | 0.70 | 4.5E-05 | 1 | UBR4,–18314; KIAA0090; MRTO4,–23014; AFAR3,37415 |
| rs1851185 | chr2:212235974 | T | 0.24 | 593 | 490 | C | 0.76 | 1745 | 1848 | 462.8 | 380.3 | 1.22 | 4.8E-05 | 1 | ERBB4 |
| rs12321966 | chr12:8592432 | T | 0.09 | 264 | 196 | G | 0.91 | 2072 | 2140 | 243.5 | 153.6 | 1.58 | 5.1E-05 | 1 | CLEC4D,26205; CLEC4E,–7607 |
| rs4805453 | chr19:34814743 | C | 0.42 | 1050 | 927 | T | 0.58 | 1286 | 1409 | 635.7 | 503.2 | 1.26 | 5.3E-05 | 1 | POP4,16196; PLEKHF1,–33423 |
| rs6901207 | chr6:3798905 | G | 0.44 | 1110 | 995 | A | 0.56 | 1228 | 1343 | 650.3 | 503.8 | 1.29 | 5.6E-05 | 1 | FAM50B,2355 |
| rs6443997 | chr3:186016225 | A | 0.06 | 106 | 156 | G | 0.94 | 2232 | 2182 | 98.0 | 159.2 | 0.62 | 6.1E-05 | 1 | VPS8 |
| rs795955 | chr12:77160181 | T | 0.40 | 883 | 993 | C | 0.60 | 1455 | 1345 | 503.7 | 621.1 | 0.81 | 6.2E-05 | 1 | NAV3,29260 |
| rs10507070 | chr12:94873188 | A | 0.17 | 451 | 359 | G | 0.83 | 1885 | 1977 | 381.6 | 285.1 | 1.34 | 6.3E-05 | 1 | CCDC38,–12629; AMDHD1; HAL,18084; LTA4H,45553 |
| rs7179849 | chr15:22589304 | T | 0.18 | 383 | 475 | C | 0.82 | 1955 | 1863 | 305.7 | 389.8 | 0.78 | 6.6E-05 | 1 | SNRPN,–30582 |
| rs7180015 | chr15:85305969 | G | 0.09 | 162 | 226 | A | 0.91 | 2176 | 2112 | 144.4 | 225.8 | 0.64 | 7.1E-05 | 1 | AGBL1 |
| rs1782 | chr6:90124434 | C | 0.12 | 311 | 240 | T | 0.88 | 2017 | 2088 | 279.7 | 197.7 | 1.41 | 7.7E-05 | 1 | GABRR2,–42748; UBE2J1,–5096; RRAGD,9878 |
| rs2362643 | chr16:68503033 | G | 0.32 | 815 | 701 | A | 0.68 | 1523 | 1637 | 578.2 | 436.4 | 1.32 | 7.8E-05 | 1 | WWP2; LOC348174,–39277 |
| rs2211871 | chr21:38744520 | G | 0.09 | 242 | 179 | T | 0.91 | 2094 | 2157 | 222.4 | 166.6 | 1.33 | 8.3E-05 | 2 | ERG |
| rs4925449 | chr22:47486086 | A | 0.08 | 170 | 237 | G | 0.92 | 2166 | 2099 | 153.4 | 207.6 | 0.74 | 8.8E-05 | 1 | FAM19A5 |
| rs175 | chr7:25000316 | C | 0.47 | 1142 | 1031 | A | 0.53 | 1194 | 1305 | 635.4 | 527.1 | 1.21 | 9.2E-05 | 1 | OSBPL3,–14031 |
| rs10760120 | chr9:99908721 | G | 0.47 | 1024 | 1143 | A | 0.53 | 1308 | 1189 | 514.3 | 646.8 | 0.80 | 9.3E-05 | 1 | NANS,23543; TRIM14; CORO2A,17575 |
| rs10489577 | chr1:231021449 | C | 0.04 | 103 | 61 | T | 0.96 | 2235 | 2277 | 100.0 | 62.3 | 1.60 | 9.5E-05 | 1 | KIAA1383,8734 |
| rs921383 | chr11:77388489 | A | 0.47 | 1172 | 1057 | G | 0.53 | 1162 | 1277 | 656.4 | 506.4 | 1.30 | 9.8E-05 | 1 | INTS4,–5124; KCTD14,15919 |
In polygenic score analyses (Figure 2), family-based results significantly predicted PGC case-control status for all thresholds, with the lowest p value of 1×10−17 (explaining 0.4% of the variance) achieved for 34,937 SNPs with p<0.2 in the family study.
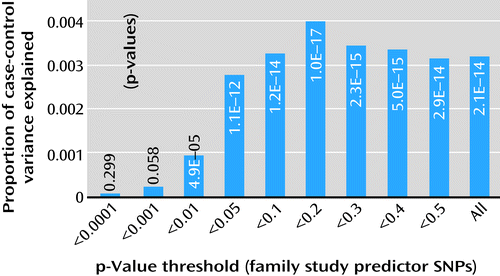
a Each bar shows the proportion of variance explained (R2) in analyses using polygenic scores (1), computed based on association test results from this family-based study, to predict the case-control status of 9,394 schizophrenia case subjects and 12,462 control subjects from the Psychiatric GWAS Consortium (PGC) GWAS (6). A subset of 112,869 family study SNPs was selected for which PGC had data (genotyped or imputed from HapMap 3 information with information content >0.9), with minor allele frequency >2% in both data sets, and correlation (r2) between SNPs <0.25. Shown below each bar is the proportion of the SNPs (rank-ordered by family study p value) used in that analysis. For each PGC subject, a polygenic score was computed by multiplying (for each SNP) the family study association test result (log[odds ratio]) by the subject’s genotype (how many of the designated test alleles the subject carried) and then summing these products across SNPs. The p value shown within each bar is from a logistic regression of PGC case-control status predicted by polygenic scores plus seven ancestry-based covariates. The R2 is the difference between Nagelkerke’s R2 for prediction using scores and covariates minus the R2 for covariates alone. (See Table S3 in the online data supplement for additional details.) The best prediction was observed when SNPs with the best 20% of p values were included. The prediction is highly significant, although with a very small proportion of total variance explained.
PGC and family study odds ratios were in the same direction for 37 of the 58 tested SNPs (one-sided binomial p=0.024) (see Table S4 in the online data supplement), or 29/45 after excluding proxy SNPs with r2<0.8 (p=0.036).
ALIGATOR analyses (see Tables S5 and S6 in the online data supplement) did not detect significant pathway effects (single pathways or excess of number of pathways) after correction for multiple testing.
Previously Documented CNV Regions
Figure 3 illustrates eight pedigrees with CNVs with previous significant evidence for association with schizophrenia (7). We observed 1q21.1 and 15q13.3 duplications segregating with schizophrenia in offspring, but only the reciprocal deletions have been strongly associated in these regions, with weaker evidence for 1q21.1 duplications (7). One of two affected offspring had an exonic NRXN1 deletion, but not the unaffected father (the mother was unavailable). For 16p11.2, duplications were observed in an unaffected mother and two of three affected children. The recruiting site reported a duplication in an unaffected sibling (not genotyped here) (34). It is unlikely that the affected father, who was deceased, carried the same rare CNV. Four cases had 22q11.2 deletions (three typical 3 Mb and one proximal 1.5 Mb), all de novo. Excluding the 15q duplication, these CNVs were seen in seven of 633 families (1.1%), compared with 1.3% of cases in a recent meta-analysis (7). No large 3q29 deletions or exonic VIPR2 duplications were observed (7).
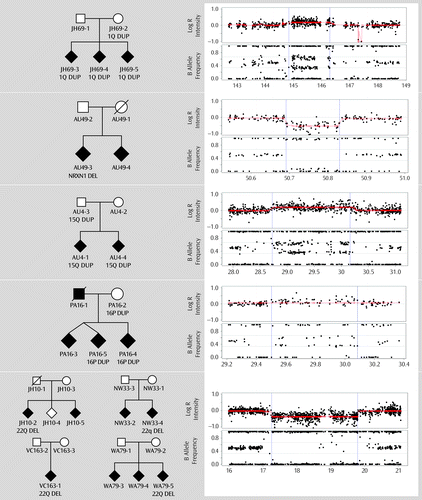
a Shown are the eight pedigrees with carriers of CNVs in five chromosomal regions with well-documented association of CNVs to schizophrenia, including 1q21.1 (typical HG18 boundaries 144.6–146.3 Mb), NRXN1 (interrupting exons of the gene, which lies on chromosome 2, 50–51.1 Mb), 15q13.3 (28.7–30.3 Mb), 16p11.2 (29.5–30.1 Mb), and 22q11.2 (17.1–20.2 Mb, or less commonly, a proximal 1.5 Mb deletion, as observed in individual NW33-4). Only genotyped offspring and their parents are shown, but all families were multiply affected. An illustrative example of each CNV is shown: the top plot shows the log(R) intensity (also known as log[R] ratio) for each probe location, with point-by-point estimates (in red) of changes in copy number (up for duplications, down for deletions) using a second algorithm (33). The bottom plot shows the B-allele frequency, i.e., where copy number=2, the designated “B” allele has 0%, 50%, or 100% of the total fluorescent intensity, but when copy number=1, only values of 0% or 100% are seen, while with copy number=3, some alleles have 33% or 67% of the total intensity, producing a distinctive pattern as shown. (Family IDs are masked.)
Discussion
Our results suggest that there is substantial overlap between the common SNPs that confer schizophrenia risk in multiply affected families and in unrelated cases, based on the highly significant polygenic score analysis: when association test results from the family study were used to weight the genotypes of PGC subjects, the resulting polygenic scores significantly differentiated case subjects from control subjects. Note that this result does not prove that there are no genetic effects that are individually stronger or more prevalent in multiply affected families.
It has been proposed that this cross-study consistency is due to a large number (perhaps many hundreds) of risk SNPs in the genome (1, 35). In very large samples, the best results will contain some true associations; for example, in the PGC two-stage analysis of single SNPs, seven chromosomal regions ultimately produced highly significant results, drawn from 58 independent SNPs in the best 53 regions of association in stage 1 (6) (most of them with consistent directions of effect in the family sample). Here, with a small predicting sample, the polygenic score analysis became significant as the proportion of best SNPs included in the analysis increased from 0.1% to 1%, but it was most significant using the best 20%, and in the PGC analysis (with a much larger predicting sample), significance continued to improve when all independent SNPs were included. This suggests that risk SNPs are distributed across the range of p values (or odds ratios), because most of them gave quite small individual effects. Polygenic score analysis cannot currently determine which SNPs are truly involved in risk. Here, network-based analyses did not further define the polygenic effect, and it is likely that an increased understanding of gene and protein functions and interactions will be needed to accomplish this.
The actual proportion of variance in PGC case-control status that could be explained was quite low (0.4%). The variance that can be explained by this type of cross-data set analysis is limited by the need to use only independent SNPs in the analysis, by the fact that GWAS assays do not provide information about all common SNPs, and by loss of information as a result of differences in genotyping methods and ancestral backgrounds of samples. Other forms of analysis suggest that common SNPs actually explain around 20%–30% of the genetic variance for schizophrenia (1, 36). Polygenic score analyses of case-control samples have predicted larger amounts of variance as the predicting sample size has increased, from around 4% with prediction and test samples with approximately 3,000 cases (1) to approximately 7% with a larger predicting sample (around 6,500 cases) and a test sample of approximately 3,000 cases. Here, we used the smaller family sample for prediction to the larger PGC case-control sample, because there is no current method for computing polygenic scores for individual subjects based on family data with some parental genotypes inferred rather than directly observed. Therefore, while our results demonstrate a highly significant overlap in common risk SNPs in these families and the PGC case sample, we cannot determine whether there is any reduction in overlap in multiplex families compared with unrelated cases.
It has been suggested that this polygenic signal could be due in part to weak correlations between common SNPs and nearby rare SNPs or structural variants with larger effects on risk (37). Most evidence does not favor this hypothesis (35); for example, we have not found single families with significant linkage signals that might be produced by rare, heritable large-effect variants. The next generation of sequencing-based studies might shed more light on the genetic effects of various types of sequence and structural variants across the full range of frequencies.
We did not observe larger effect sizes of single SNPs in these multiply affected families than have been reported in case-control samples (www.genome.gov/gwastudies, accessed May 7, 2011). Because exonic deletions in NRXN1 are the only single-gene mutations shown to be associated with large increases in schizophrenia risk (approximately eightfold) (7), we were interested to note that several SNPs with low p values were in or near genes with related functions involving brain development and neuronal cell adhesion and signaling (CNTNAP5, CADM2, ERRB4, PPFIA2, PTPRN2, CLEC4D/E, AMIGO3, and CNTN5 for all ancestries). However, we did not detect statistically significant evidence for association of any defined pathway after correcting for multiple testing of pathways. This could be due to lack of statistical power from the relatively small sample size or because the pathophysiological mechanisms underlying schizophrenia risk are not adequately captured by current pathway definitions.
Five rare CNVs are strongly associated with schizophrenia, and three of them (16p11.2 duplications, 22q11.2 deletions, and NRXN1 exonic deletions) were observed here, along with duplications that are reciprocal to associated deletions of 1q21.1 and 15q13.3; there is some evidence for association of 1q21.1 duplications, but not for 15q13.3 duplications (7). The total frequency of these CNVs (excluding the 15q13.3 duplication) was similar to that observed in previously reported case samples. The family data provide several insights. First, the possibility of a de novo (nontransmitted) 22q11.2 deletion should not be ignored in multiply affected families—indeed, the prevalence of these deletions was similar to that reported in large samples with primarily nonfamilial cases (7). There must have been other genetic or nongenetic risk factors in these families, but it is not known whether their effects were limited to the siblings without a 22q11.2 deletion or whether they also influenced the emergence of the schizophrenia phenotype in the carrier, given that schizophrenia develops in only ∼30% of 22q11.2 carriers. Second, two transmitted CNVs (16p11.2 duplications and a NRXN1 deletion) failed to segregate perfectly with schizophrenia within the family, suggesting again that other risk factors were present.
Conclusions
This GWAS of multiply affected families produced significant support for a polygenic model that posits that multiple common SNPs confer part of the genetic risk of schizophrenia, with a significant overlap between common risk SNPs in multiply affected families and samples of unrelated case subjects. Significant association was not detected for any single SNP, which is consistent with the relatively small sample size, but for the most significant SNPs in the large PGC GWAS analysis, the direction of effect was the same in both samples for a significant excess of SNPs. Several of the “top SNPs” in the family study were in genes related to neurodevelopment, but no statistically significant evidence was observed for association of currently defined gene pathways. Rare CNVs were observed in regions with strong previously documented association with schizophrenia, but with variable patterns of segregation. This should serve as a reminder that we still know relatively little about the distribution of these CNVs in the entire population (e.g., in individuals with no or only mild cognitive problems) or about the reasons for the emergence of schizophrenia in only a minority of carriers, so great caution is required in genetic counseling and prediagnosis.
1 : Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460:748–752Crossref, Medline, Google Scholar
2 : Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460:753–757Crossref, Medline, Google Scholar
3 : Common variants conferring risk of schizophrenia. Nature 2009; 460:744–747Crossref, Medline, Google Scholar
4 : The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry 2011; 70:35–42Crossref, Medline, Google Scholar
5 : Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol Psychiatry 2011; 16:429–441Crossref, Medline, Google Scholar
6 : Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43:969–976Crossref, Medline, Google Scholar
7 : Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry 2011; 168:302–316Link, Google Scholar
8 : Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet 1998; 20:70–73Crossref, Medline, Google Scholar
9 : No evidence for involvement of KCNN3 (hSKCa3) potassium channel gene in familial and isolated cases of schizophrenia. Eur J Hum Genet 1999; 7:247–250Crossref, Medline, Google Scholar
10 : Genetic study of dopamine D1, D2, and D4 receptors in schizophrenia. Psychiatry Res 1994; 51:215–230Crossref, Medline, Google Scholar
11 : Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics 1997; 43:1–8Crossref, Medline, Google Scholar
12 : Genome scan of schizophrenia. Am J Psychiatry 1998; 155:741–750Abstract, Google Scholar
13 : A genome-wide autosomal screen for schizophrenia susceptibility loci in 71 families with affected siblings: support for loci on chromosome 10p and 6. Mol Psychiatry 2000; 5:638–649Crossref, Medline, Google Scholar
14 : Irish study on high-density schizophrenia families: field methods and power to detect linkage. Am J Med Genet 1996; 67:179–190Crossref, Medline, Google Scholar
15 : A two-stage genome scan for schizophrenia susceptibility genes in 196 affected sibling pairs. Hum Mol Genet 1999; 8:1729–1739Crossref, Medline, Google Scholar
16 : Genomewide linkage scan of schizophrenia in a large multicenter pedigree sample using single nucleotide polymorphisms. Mol Psychiatry 2009; 14:786–795Crossref, Medline, Google Scholar
17 : Multicenter linkage study of schizophrenia candidate regions on chromosomes 5q, 6q, 10p, and 13q: schizophrenia linkage collaborative group III. Am J Hum Genet 2000; 67:652–663Crossref, Medline, Google Scholar
18 : No major schizophrenia locus detected on chromosome 1q in a large multicenter sample. Science 2002; 296:739–741Crossref, Medline, Google Scholar
19 : Multicenter linkage study of schizophrenia loci on chromosome 22q. Mol Psychiatry 2004; 9:784–795Crossref, Medline, Google Scholar
20 : Continuity and discontinuity of affective disorders and schizophrenia. Results of a controlled family study. Arch Gen Psychiatry 1993; 50:871–883Crossref, Medline, Google Scholar
21 : Diagnostic accuracy and confusability analyses: an application to the Diagnostic Interview for Genetic Studies. Psychol Med 1996; 26:401–410Crossref, Medline, Google Scholar
22 : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38:904–909Crossref, Medline, Google Scholar
23 : A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 1999; 65:1170–1177Crossref, Medline, Google Scholar
24 : Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered 2008; 66:87–98Crossref, Medline, Google Scholar
25 : Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet 2003; 73:1016–1026Crossref, Medline, Google Scholar
26 : Association of synapsin 2 with schizophrenia in families of Northern European ancestry. Schizophr Res 2007; 96:100–111Crossref, Medline, Google Scholar
27 : Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 2009; 85:13–24Crossref, Medline, Google Scholar
28 : A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 2009; 84:210–223Crossref, Medline, Google Scholar
29 : Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455:237–241Crossref, Medline, Google Scholar
30 : Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull 2009; 35:851–854Crossref, Medline, Google Scholar
31 : Large recurrent microdeletions associated with schizophrenia. Nature 2008; 455:232–236Crossref, Medline, Google Scholar
32 : PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007; 17:1665–1674Crossref, Medline, Google Scholar
33 : Stochastic segmentation models for array-based comparative genomic hybridization data analysis. Biostatistics 2008; 9:290–307Crossref, Medline, Google Scholar
34 : Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry 2009; 66:947–956Crossref, Medline, Google Scholar
35 : Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol 2011; 9:e1000579Crossref, Medline, Google Scholar
36 : Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatry 2012; 17:474–485Crossref, Medline, Google Scholar
37 : Rare variants create synthetic genome-wide associations. PLoS Biol 2010; 8:e1000294Crossref, Medline, Google Scholar

