
Copy Number Variations in Schizophrenia: Critical Review and New Perspectives on Concepts of Genetics and Disease
Abstract
Objective
Structural variations of DNA, such as copy number variations (CNVs), are recognized to contribute both to normal genomic variability and to risk for human diseases. For example, schizophrenia has an established connection with 22q11.2 deletions. Recent genome-wide studies have provided initial evidence that CNVs at other loci may also be associated with schizophrenia. In this article, the authors provide a brief overview of CNVs, review recent findings related to schizophrenia, outline implications for clinical practice and diagnostic subtyping, and make recommendations for future reports on CNVs to improve interpretation of results.
Method
The review included genome-wide surveys of CNVs in schizophrenia that included one or more comparison groups, were published before 2009, and used newer methods. Six studies were identified.
Results
Despite some limitations, these initial genome-wide studies of CNVs provide replicated associations of schizophrenia with rare 1q21.1 and 15q13.3 deletions. Collectively, the results point to a more general mutational mechanism involving rare CNVs that elevate risk for schizophrenia, especially more developmental forms of the disease. Including 22q11.2 deletions, rare risk-associated CNVs appear to account for up to 2% of schizophrenia.
Conclusions
The more penetrant CNVs have direct implications for clinical practice and diagnostic subtyping. CNVs with lower penetrance promise to contribute to our genetic understanding of pathogenesis. The findings provide insight into a broader neuropsychiatric spectrum for schizophrenia than previously conceived and indicate new directions for genetic studies.
In the ongoing search for genetic origins of schizophrenia, most of the focus has been on changes in DNA sequence that may elevate risk for the disorder. However, genomic copy number variations (CNVs) are increasingly recognized to contribute to risk for human diseases (1–3). One of the most exciting recent discoveries about the human genome is that, in addition to variations in sequence, such as single nucleotide polymorphisms (SNPs), individuals have variations in genomic structure (4–6). Structural variants, mainly CNVs involving loss (e.g., deletions) or gain (e.g., duplications) of up to several million base pairs of DNA sequence, are estimated to constitute upward of 5% of the human genome (4, 7, 8). CNVs can alter gene dosage and may involve multiple genes and/or regulatory regions (Figure 1). In general, CNV deletions show higher penetrance (more severe phenotype) than duplications and larger CNVs often have higher penetrance and/or more clinical features than smaller CNVs. It is now apparent that structural variants contribute to normal variability, disease risk, and developmental anomalies as well as acting as a major mutational mechanism in evolution (8, 9).
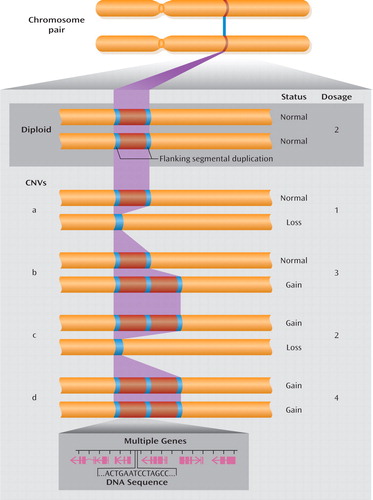
aFlanking segmental duplications increase the risk for structural genomic changes, such as CNVs. Normal diploid status is shown in the example at the top. Examples of CNVs (a to d, below) show the change in copy number in various configurations. A simple loss (example a) is often called a microdeletion and a simple gain (example b) a microduplication. While CNVs associated with genomic disorders are more prone to higher mutation rates because of the highly identical sequences in segmental duplications, most CNVs in the genome do not arise from events mediated by segmental duplication. CNVs may involve no, one, or multiple genes (genomic extent including exons and introns shown in magenta).
Technological advances have driven much of the research on CNVs and allowed detection of smaller and smaller CNVs (1, 10, 11). As with most discoveries, however, those that are the most readily detectable and have the most severe effects are reported first. Thus, although the vast majority of CNVs are inherited, CNVs that have newly occurred as de novo (spontaneous) mutations have more readily been implicated in diseases. A significant part of the CNV-disease connection involves a new category of genetic diseases called genomic disorders (3, 12). The CNVs associated with genomic disorders involve microdeletions and microduplications typically too small to be detected by standard karyotype. These often arise as de novo mutations mediated by flanking segmental duplications (Figure 1), also known as low-copy-repeat sequences, comprising nearly identical DNA sequences (>90% nucleotide similarity) and involving unequal crossing over in meiosis (3, 12). The most common genomic disorder, 22q11.2 deletion syndrome, has an established association with schizophrenia (13). Individuals with 22q11.2 deletions have a 20-fold increase in risk for schizophrenia and account for about 0.9%–1.0% of schizophrenia in the general population (13). Recent reports (14–17) suggest that CNVs at other loci may also be associated with schizophrenia. Understanding how to interpret these findings and their implications is the goal of the current review. We begin with a brief overview of CNVs, then review the recent CNV findings related to schizophrenia, and conclude by outlining the implications of this research for clinical practice and diagnostic subtyping, making recommendations for future reports.
Overview of Copy Number Variations
Detection, Mechanisms, and Expression
CNVs may be detected by targeted or genome-wide methods. The latter include karyotype with a lower resolution of at best 5–10 megabases (Mb) of DNA (a typical band of a chromosome) and multiple genome-wide DNA microarrays with coverage and resolution that vary according to the individual technology platform, probe set, and methodology. Most microarrays use SNP-based or array comparative genomic hybridization (aCGH) methods. The former require algorithms to assist in determining copy number, and the latter rely on one or more "hybridization reference" genomes, the choice of which can influence results. These methods vary in their resolution and ability to determine the break points and extent of individual CNVs. There is also rapid progress to using whole-genome ("next-generation") sequencing methods (18). Targeted methods include those often used in clinical laboratories that detect microdeletions or microduplications associated with genomic disorders, such as fluorescence in situ hybridization (FISH) using specific probes and molecular cytogenetic methods, and microarrays using various probe-screening sets that offer increasingly extensive coverage. Quantitative polymerase chain reaction (qPCR) is another targeted method often used to confirm a CNV identified by using array-based methods and to quickly screen large sample sets (1).
Much remains to be discovered about the mechanisms giving rise to CNVs and the pathways from CNVs to phenotypic expression recognizable as illness. Repeated elements in the genome have been implicated in many but not all CNV break points (8, 19). Mechanisms include meiotic unequal crossing over, or nonallelic homologous recombination, mediated by flanking repeated sequences or segmental duplications, and nonhomologous DNA repair mechanisms such as nonhomologous end joining or microhomology-mediated end joining (19). If DNA repair mechanisms during mitotic divisions of germ cells are involved, this could lead to new mutations arising in sperm through adulthood, possibly associated with late paternal age, or in maternal grandchildren, given the timing of these divisions in fetal development for females (19).
While CNVs make a significant contribution to observable variability between individuals, there is not always a straightforward correlation between gene dosage and expression (20, 21). Larger CNVs on average will encompass more genes, but they may also contain regulatory elements for genes and they can in addition exert effects on chromatin structure. Therefore, CNVs can have long-distance effects on expression of genes in neighboring chromosomal regions, even up to 1 Mb away from a particular structural variant (20–22).
Association With Disease
There are numerous complexities in phenotypic categorization, CNV detection and characterization, and statistical analyses that need to be considered in CNV-disease associations. Schizophrenia has strong evidence for neurodevelopmental origins (23, 24). Thus, schizophrenia may be expected to have CNV associations similar to those in disorders such as autism, where specific CNVs and conceptually similar but microscopically visible chromosomal anomalies detectable by karyotype are present in a substantial minority (approximately 10%) of cases (25–28). Hallmarks of developmental changes, e.g., mental retardation, birth defects, dysmorphic facies, and/or childhood-onset schizophrenia, could indicate individuals with schizophrenia for whom the a priori likelihood of having associated CNVs may be elevated. Sampling strategies can significantly influence the observed prevalence of CNVs in both individuals with the disorder and comparison subjects. In general, no or low prevalence in comparison subjects would be expected for highly penetrant CNVs and higher prevalence in comparison subjects may be expected with less-penetrant CNVs. Likewise, familial forms of schizophrenia may be less commonly associated with certain CNVs than "sporadic" forms of illness, depending on the likelihood of de novo mutations and penetrance of the disease for the specific CNV. Ideally, one would be able to assess all individuals with rare CNVs of interest to determine the full phenotypic spectrum and overt or cryptic relatedness to others who carry the same CNV.
In contrast to SNPs, for which one can expect a 99% sensitivity and specificity of detection of the targets on the microarray, the complex nature of CNVs (Figure 1) and related experimental issues do not allow the same level of confidence for CNV detection (1). Uncertainty about the presence, extent, and break points of CNVs affects analyses of the genes that may be involved. In general, with current commercial microarrays large CNVs (>100 kb) are more reliably detectable, have greater overlap between platforms, and are easier to assign pathological status, particularly if they arise as de novo mutations.
Recurrent de novo CNV events and/or multiple rare CNVs that show convergence on a single gene or set of genes (or networks or pathways of genes) strengthen the association with disease. Although SNP-based arrays have poor coverage of segmental duplications, large CNVs in intervals between such complex repeat regions are readily detectable (29). Sources of possible laboratory- and/or array-based variability include variable probe performance and measurement error, batch effects and "noise" from cell line artifacts, and/or poor-quality DNA samples. Platforms vary with respect to coverage across the genome; more recent arrays with 1 million or more SNPs increase detection down to CNVs spanning less than 20 kb. Smaller CNVs, which constitute the majority of those detected by using SNP-based platforms, are usually inherited (11). It is as yet unclear whether smaller CNVs will have properties similar to those of large CNVs with respect to association with diseases like schizophrenia.
Common CNVs (allele frequency >5%) have properties similar to those of SNPs, comprise the majority of CNV differences between individuals, and are almost always inherited (8, 11). Most are in strong linkage disequilibrium with SNPs and thus may be analyzed with standard genetic association methods. Genuine associations of CNVs with disease may be difficult to determine for rare events with low prior probability, however. Accurate estimates of such associations will depend on an individual with a rare CNV being reported only once or, if included in different studies, clearly demarcated as such. Parental data, i.e., phenotypes and genotypes, are valuable, although not always available given the late age at onset of schizophrenia. Data on family members carrying the CNV provide information relevant to estimates of penetrance and variable expression. Case-control studies rely on the comparison groups being free of disease and unrelated to the affected individuals. The relatively high prevalence of schizophrenia in the general population and reduced penetrance and/or variable expression of genetic variants present further challenges to interpreting CNV results. Given all of these factors, prevalence figures and odds ratios will be very approximate estimates at this early stage of CNV studies. Independent replication is essential.
Review Methods
For this review, we included genome-wide surveys of CNVs in schizophrenia that included one or more comparison groups and were published before Jan. 1, 2009. We excluded studies using earlier aCGH methods based on bacterial artificial chromosomes, as these platforms tend to lack precision because of the large size of the probes. We examined each report, all supplemental materials, and where possible, other reports of the samples and individuals with major CNVs. We made every attempt to determine subjects that overlapped between studies to supplement sampling descriptions and to be better able to discuss independent replication of CNV results. We abstracted methodological data, including ascertainment and study sites, phenotype, array platform, and selected recurring CNVs, focusing on the salient issues to consider in study design and how best to interpret emerging CNV data.
Results
We reviewed six genome-wide studies reporting on CNVs in schizophrenia, all of which were published in 2008. Table 1 presents the data on the major recurrent CNVs reported in these studies, with further details of sampling issues summarized in footnotes. In the four smaller studies (14, 15, 30, 34) we observed no overlap of study groups. The largest two studies, however, involved individuals originally collected for various purposes at multiple sites in several countries, predominantly in northern Europe (16, 17). Data on subjects from one of these sites (Aberdeen, Scotland) were included in both studies (16, 17), complicating evaluation of the results. Aberdeen had the highest number of subjects with 22q11.2 deletions and 1q21.1 deletions (Table 1). However, even if this site is excluded, the studies appear to support a previously established association with 22q11.2 deletions and two new associations: rare 1q21.1 and 15q13.2–15q13.3 deletions (Table 2). Results suggesting more CNVs overall in subjects with schizophrenia than in comparison subjects (so-called CNV load or burden) are more challenging to adjudicate given significant methodological variability, e.g., inclusion (or not) of groups that were filtered for karyotypic abnormalities (14), which may skew results. Results for smaller CNVs involving single genes, while requiring further independent reports, are beginning to provide evidence of convergence on genes and pathways of interest for neurodevelopment.
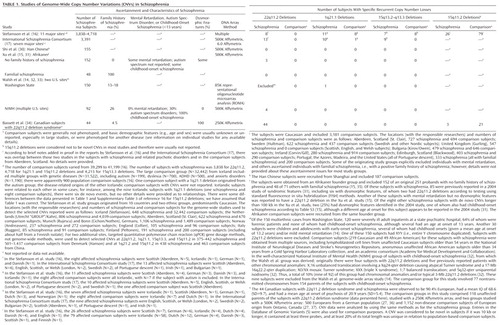 |
Newly Identified Major CNVs and Lower-Penetrance CNVs Associated With Schizophrenia
Up to 18 subjects with schizophrenia who had 1q21.1 deletions, originating from eight sites, were reported in three studies involving over 6,000 patients with schizophrenia (Table 1) (14, 16, 17). There were up to 15 schizophrenia subjects from 10 originating sites reported with 15q13.3 deletions in the studies reported by Stefansson et al. (16) and the International Schizophrenia Consortium (17) (Table 1). Both studies indicated that the proportion of subjects with schizophrenia and these CNVs was significantly different from the proportion of subjects with these CNVs in the comparison groups used. As expected for case-control studies with no capability of following up subjects, limited data are available on the phenotype or de novo versus inherited status of the individuals from either the schizophrenia or comparison groups with these deletions.
Results for the 15q11.2 locus are less clear. In one study (16), the 471-kb CNV microdeletion at 15q11.2 was reported, in fairly large numbers, in both the schizophrenia subjects and comparison subjects with unspecified disorders, including autism (Table 1). While the difference was reportedly significant, the effect size appeared smaller than that for the 1q21.1 and 15q13.3 deletion CNVs (16). A later publication that included data from the International Schizophrenia Consortium study supported the possibility that CNVs at the 15q11.2 locus are risk factors for schizophrenia with reduced penetrance (47). Other recurrent CNVs flanked by segmental duplications previously reported in autism and/or developmental delay (25–27, 48–50) that may also be risk factors for schizophrenia with reduced penetrance include 400–600-kb 16p11.2 deletions and duplications (14, 16) and duplications at 16p13.1 (1.16 Mb) (47, 51), 1q21.1 (1.4 Mb) (16, 51), and 22q11.2 (3–4 Mb) (47); the latter two are reciprocal to higher-penetrance deletions at the same loci.
Overall Prevalence of CNVs in Schizophrenia
To address the hypothesis that a general mechanism of increased spontaneous copy number mutations could be involved in causing schizophrenia, one would like to know the answer to the question, Is the overall prevalence of CNVs higher in schizophrenia than in the general population? Some of the studies reviewed suggest that this is the case, but to interpret the findings reported one needs to consider the methods used. As may be expected for such different sampling, molecular, and analytic methods (Table 1), there were no consistent CNV prevalence data reported in all six studies that would allow direct comparisons, for either the schizophrenia or the comparison groups; thus, they are considered individually.
The International Schizophrenia Consortium study considered CNVs that were more than 100 kb long and found at a frequency of less than 1% in the comparison groups used (17). The CNVs discovered were 1.15-fold more common in the total schizophrenia group than in the total comparison group, in which the base rate was 0.99 CNVs per subject. Multiple subanalyses were presented. A 1.67-fold increase in schizophrenia over the total comparison group was the largest reported, obtained by restricting the analysis to loss CNVs more than 500 kb in size that had the lowest base rate (0.03) per subject in the comparison group (17). Not surprisingly, especially given that the large, gene-rich 22q11.2 deletions, as well as 1q21.1 and 15q13.3 deletions, were included (Table 2), the schizophrenia group was reported to have on average 3.57-fold more genes involved in CNVs than the comparison group. Individual genome-wide CNV data were not provided.
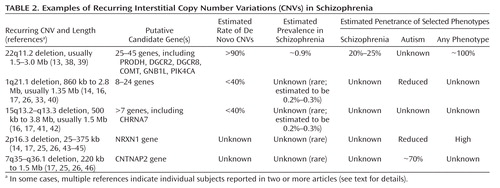 |
In some cases, multiple references indicate individual subjects reported in two or more articles (see text for details).
The multisite study of Stefansson et al. (16) covered 66 CNVs that had been identified as de novo events in genome-wide surveys of Icelandic samples of parents and offspring from 7,718 parent-offspring pairs or trios. Samples from this genetic isolate had been assembled for various genetic studies of medical and psychiatric disorders, such as autism, but excluding schizophrenia. This strategy thus enhanced for recurrent CNVs with variable expression. Genome-wide CNVs, except for the 66 selected CNVs, were not reported, precluding comparisons with data from other populations.
Walsh et al. (14) considered CNVs greater than 100 kb that had not been described in the literature or in public databases as of November 2007 and that affected genes. They reported a threefold greater proportion with these CNVs in their forensic and adolescent groups with schizophrenia (22 of 150 subjects) than in the composite comparison subjects used (13 of 268 subjects). Inheritance or de novo status of the CNVs was unknown. The schizophrenia group excluded subjects with 22q11.2 deletions but included a subject with a sex chromosome anomaly, 47,XYY. This report also included a study using a different array platform and another group comprising 83 subjects with childhood-onset schizophrenia. This group excluded nine additional subjects with chromosomal anomalies or 22q11.2 deletions (32). For the same categories of CNVs, this group showed a 2.6-fold greater proportion (23 of 83) than did the parental comparison subjects used (10 of 77). It is not clear why the proportion of individuals studied who had CNVs longer than 100 kb was lower in the comparison group for the forensic and adolescent subjects than in both the subjects with childhood-onset schizophrenia and the parental comparison group, but array differences and small study group sizes could be factors. Two (2.4%) of the CNVs in patients with childhood-onset schizophrenia were de novo changes. The expected de novo CNV mutation rate is approximately 1% at this resolution of analysis in comparison populations. Some individual genome-wide CNV data were provided.
Xu et al. (15) considered rare de novo CNVs of all sizes and prevalences and reported an eightfold greater number of these CNVs per subject in the Afrikaner subjects with sporadic schizophrenia (15 of 152), including three with 22q11.2 deletions, than in the comparison group used (two of 159). In contrast, there was a barely significant difference between groups with respect to inherited CNVs (46 of 152 versus 32 of 159). With few exceptions, individual data on genome-wide CNVs were not provided, precluding comparisons with data from other populations.
Using a study group of similar size to the groups in the studies by Walsh et al. (14) and Xu et al. (15), Shi et al. (30) reported no significant difference in the proportion of subjects with rare (<1%) CNVs larger than 100 kb between individuals with schizophrenia (109 of 155) and a comparison group (132 of 187). Results were similar whether or not the CNVs involved genes (30). No individual genome-wide CNV data were provided.
Our study (34) focused on subjects with 22q11.2 deletions and included all other CNVs in the analyses. There were no significant differences between subjects with and without schizophrenia or between those with 22q11.2 deletions and unaffected parents on any CNV-related measure, regardless of size, rarity, or de novo status. The genome-wide CNV results were also similar to comparable CNV data available from large general population samples (34). Moreover, there was no evidence of an increase in de novo CNVs as an underlying mechanism for occurrence of the 22q11.2 deletion. This study provided individual CNV data for all subjects studied.
CNVs Implicating Individual Genes
In addition to the major CNVs just discussed, the studies reviewed provided some initial evidence for recurring smaller CNVs that may implicate individual candidate genes for schizophrenia (Table 2). Some methodological issues, however, obscure the published findings.
NRXN1
Three of the genome-wide studies included individuals with schizophrenia and 2p16.3 CNVs involving the NRXN1 gene, which encodes neurexin, a scaffolding protein. Walsh et al. (14) reported monozygotic twins concordant for childhood-onset schizophrenia and a 2p16.3 loss CNV. A supplementary figure in the findings of the International Schizophrenia Consortium included five subjects with schizophrenia and six comparison subjects with 2p16.3 CNVs (17). In a previous article, the consortium also reported a Bulgarian affected sibling pair with a similar CNV at one of the study sites (43). In the study by Stefansson et al. (16), the 2p16.3 loss CNV was one of the initial 66 de novo CNVs examined, which had been identified in a proband with autism (44). In total, there appear to have been 12 subjects with CNVs involving NRXN1 in that study, three of whom appear to have been previously reported in a Utrecht study group with schizophrenia (45). The multisite results were reported in a separate publication (44) that produced a significant result when it focused on a subset of affected subjects with CNVs (six with losses, one with a gain) that disrupted exons in NRXN1 and a comparison group that excluded subjects ascertained with major psychiatric illnesses.
CNTNAP2
In a supplementary figure, the International Schizophrenia Consortium (17) reported three subjects with schizophrenia who had 7q35 loss CNVs and one comparison subject with a nonoverlapping, smaller 7q35 loss CNV involving the CNTNAP2 gene, which codes for contactin-associated protein 2. One of these three subjects with schizophrenia and a 220-kb 7q35 deletion CNV may have been included previously in the Friedman et al. study of CNTNAP2 gene dosage, which used subjects from Utrecht (46). A small, 80-kb 7q35 loss CNV involving this gene was reported in a nonpsychotic subject with a 22q11.2 deletion (34). The other multisite study reported a 7q35 loss CNV affecting the CNTNAP2 gene as one of the 66 de novo CNVs in an Icelandic proband with an unspecified condition (16). This group has not subsequently reported results for this CNV.
Discussion
Despite some methodological issues, the recent genome-wide CNV studies reviewed provide evidence that certain recurring CNVs are associated with schizophrenia. This confirms a mechanism of genetic mutation for schizophrenia that has implications in both clinical and research domains. Individually, these CNVs are rarely associated with susceptibility for schizophrenia (Table 2). The total number of individuals with schizophrenia and these CNVs reported to date is small, and any effect sizes calculated must be considered very exploratory (52). Nevertheless, the data suggest that, in addition to 22q11.2 deletions, rarer 1q21.1 and 15q13.3 deletions are associated with schizophrenia (52). Although further well-designed studies (Table 3) will be needed to determine true prevalence, the available data suggest that these three deletions may account for about 1%–2% of all cases of schizophrenia. Studies published in 2009 provide support for these deletions and several large lower-penetrance CNVs but not for a general increased prevalence of CNVs in schizophrenia (47, 51).
 |
In some cases, multiple references indicate individual subjects reported in two or more articles (see text for details).
Major Recurrent CNVs Associated With Schizophrenia
22q11.2 deletions
A consistent finding of these CNV studies was the absence of any 22q11.2 deletions in any of the comparison groups investigated, supporting the generally pathogenic nature of these deletions and the high penetrance of observable phenotypes (Table 2). The phenotypes associated with 22q11.2 deletions are known to be highly variable in number and severity, comprising congenital and later-onset physical and neuropsychiatric conditions, including epilepsy in a minority and learning difficulties in the majority. What new data do these genome-wide CNV studies offer on the well-established association of 22q11.2 deletions and schizophrenia? The prevalence of 22q11.2 deletions in schizophrenia in the multisite studies (16, 17) was about one-tenth as high (about 0.2%–0.4%) as that in the Afrikaner study group (2%) (31) used in the Xu et al. study (15) or in the original group of subjects with childhood-onset schizophrenia (4%) (32) from which one of the Walsh et al. study groups was derived (14). Estimates suggest that the overall prevalence of 22q11.2 deletions in schizophrenia is 0.9%–1% (13). Reasons for these varying prevalence estimates may involve the number and selection of subjects (13). Truly inclusive population-based prevalence samples of schizophrenia are difficult to obtain, and it is unclear whether any of the originating groups in the studies reviewed could be considered representative of a general schizophrenia population. With notable exceptions, e.g., the Aberdeen study group (16, 17), many of the originating subject groups for the consortium studies may have implicitly or explicitly excluded subjects with dysmorphic features, birth defects, learning difficulties, or known genetic syndromes. Age may have been another factor; the average age at death in 22q11.2 deletion syndrome is in the 40s (53). These sampling issues may similarly have affected the observed prevalence of other CNVs.
1q21.1 deletions
The data available suggest that the approximate prevalence of 1q21.1 deletions in schizophrenia is about 0.2%–0.3%, comparable to prevalence estimates in mental retardation (33, 40). Genome-wide studies indicate that 1q21.1 deletions may be de novo or inherited and that the phenotype is variable, including mental retardation, autism, attention deficit hyperactivity disorder (ADHD), and seizures (33, 40). Among 5,218 patients with mental retardation and congenital anomalies at 12 centers, there were 25 with 1q21.1 deletions (33). Although they may be inherited from apparently unaffected parents (33), no 1q21.1 deletions were found in 4,737 comparison subjects (33), a finding consistent with results for the schizophrenia CNV studies (Table 2) and indicating these rare deletions are often pathogenic in nature. This is supported by another study of 16,557 patients from multiple centers whose samples were sent to a single clinical laboratory, of whom 21 showed 1q21.1 deletions (40). No schizophrenia was reported in either study, but few adults were included. Minor dysmorphic features were variable, with a suggestion that microcephaly may be associated (33, 40). Few such details were available for the estimated 18 subjects in the schizophrenia studies (14, 16, 17).
As with 22q11.2 deletions (34), the phenotype of 1q21.1 deletions showed no correspondence to the extent of the deletion. The 1q21.1 deletion region is highly complex, with at least four large segmental duplications (4). The 1q21.1 deletion associated with a genomic disorder appears to most commonly involve the 1.35-Mb region between two of these sequences and less commonly a larger deletion that extends proximally (4, 33, 40), similar to those in the schizophrenia CNV studies (16, 17) and confirmed for one subject (14, 33).
15q13.3 deletions
The 15q13.3 deletions associated with genomic disorders are, most commonly, the 1.5-Mb region between two large segmental duplications (breakpoints 4 and 5) and, less frequently, a larger, 3.8-Mb deletion extending more proximally to another segmental duplication (breakpoint 3) (41, 54). These deletions have now been reported in as many as 15 subjects with schizophrenia (Table 2), including some with mental retardation and/or seizures or epilepsy (16, 17), nine subjects with mental retardation, including one with autism (41), 14 children with cognitive and autistic or attention disorders and six transmitting parents (42), and 14 subjects with idiopathic generalized epilepsy, including three with mental retardation (54). Subject overlaps between some of these studies are possible. Although the data are insufficient, there is no evidence that phenotypic severity corresponds to the extent of the deletion. Where assessed, many individuals with 15q13.3 deletions were noted to have mild and variable dysmorphic features (41). The preliminary prevalence data available suggest that these CNVs are rarer in schizophrenia or mental retardation (about 0.2%–0.3%) than they are in epilepsy (about 1%) (54). Notably, the approximate prevalence of 15q13.3 deletions in idiopathic generalized epilepsy is about the same as that of 22q11.2 deletions in schizophrenia, each CNV representing the most prevalent major risk factor identified to date for the respective complex disorder (13, 54).
Smaller Individual CNVs Implicating Specific Genes
Although the data are preliminary and involve few individuals, CNV studies provide some evidence to support the neurexin superfamily, specifically the NRXN1 and CNTNAP2 genes, as potentially important in schizophrenia. These genes cover large genomic extents, over 1 Mb for NRXN1 and over 2 Mb for CNTNAP2. Recurring CNVs may more likely affect such genes than genes of smaller size. Altogether, there appear to be 18 or 19 individual subjects with schizophrenia reported in a total of six articles to have 2p16.3 loss CNVs involving NRXN1 (44), providing some evidence that the NRXN1 gene may be involved in schizophrenia. As is the case with the larger CNVs associated with schizophrenia, the phenotype of CNVs involving these genes embraces other conditions, including autism and epilepsy (25–27, 46). In the case of CNTNAP2, Tourette's syndrome, obsessive-compulsive disorder, and ADHD may also be part of the expression profile (55, 56). The relevance of these findings awaits further data.
Limitations and Recommendations for Future CNV Studies
Relatively few investigators have yet reported their complete CNV data sets or released their raw data, minimizing the opportunity for comparing and contrasting data or for meta-analyses. As a result, some commonalities (or differences) in the data may be missed. We anticipate that the results presented are just the beginning of a rich literature that will broaden our understanding of structural variants as risk factors for schizophrenia and their possible role in genetic interactions.
We propose some recommendations (Table 3) that will help in the evaluation of CNV results and appreciation of their implications. Methodological and reporting issues significantly limit the interpretations possible from the genome-wide CNV studies reported to date. This review highlights ascertainment and phenotypic details and subject overlap that hampered adjudication of results. More rigorous phenotyping is key to genetic discoveries and to understanding implications of potential discoveries, including those related to CNVs (1, 2, 57). As for most mutations, expression associated with CNVs is variable and severe phenotypes are usually the first to be discovered (Figure 2). Early penetrance estimates for specific CNVs may therefore be higher than those available after further study, and more low-penetrance CNVs will be reported over time (47, 51). Apart from 22q11.2 deletions (34), little has been reported about the chromosomal parent-of-origin status of de novo deletions. This may be particularly important for imprinted regions, such as 15q11–q13, where a specific phenotype may be observed only in a maternally (or paternally) transmitted chromosome. There is a paucity of data on parents and other family members of individuals reported to have these CNVs, leaving the inheritance or de novo status and full range of expression largely unknown. Differing methods were used to determine rates of novel CNVs, de novo CNVs, rare CNVs, and genes, including CNS genes, that were disrupted, involved, and/or affected (Table 1). Differences in array platforms and algorithms used between study groups and between studies further limit interpretation of results. With few exceptions (14, 34), there were no or only limited validation studies of CNVs (Table 3) reported as recurring and/or of interest. The studies to date mostly involve Caucasians, and the effects of ethnicity on the prevalence of CNVs are largely unknown (4; also see the Database of Genomic Variants: http://projects.tcag.ca/variation/). Despite the limitations, however, the replicated findings provide new perspectives for the genetics of schizophrenia and genetic concepts of neuropsychiatric disease, and they have important clinical implications.
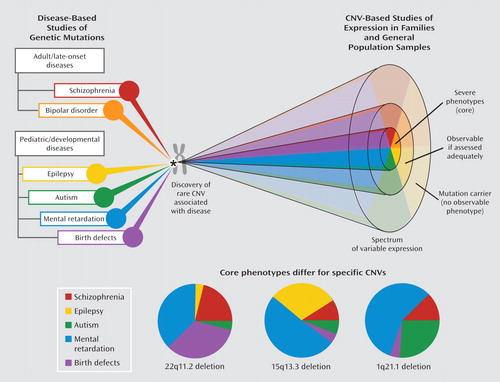
aThe top diagram illustrates the discovery of rare CNVs, which may be discovered in studies of both adult-onset and developmental diseases; studying individual CNVs can delineate the spectra of their respective phenotypic expressions. The charts below represent three CNVs having elevated prevalences in schizophrenia. These indicate the most common core phenotype of ascertainment based on current knowledge; the expression spectrum may change as more data accumulate. Proportions would differ if combined phenotypes of ascertainment (e.g., schizophrenia and mental retardation) were considered.
Clinical Relevance of Major CNVs Associated With Genomic Disorders
What is the general relevance of these CNV findings for clinicians? The 22q11.2, 1q21.1, and 15q13.3 deletions are the only major genetic risk factors identified to date for schizophrenia—and for which clinical genetic assessment and testing are widely available. The relatively modest prevalence of these anomalies suggests to us that, at this time, expensive genomic investigations are not warranted for all individuals with schizophrenia. Clinicians should, however, have a raised index of suspicion for genomic disorders or de novo CNVs in individuals with schizophrenia who have significant learning difficulties, dysmorphic facial features, birth defects, and/or unprovoked seizures (58). Family history, e.g., in offspring, of birth defects, dysmorphic features, developmental delay, and/or autism should also prompt consideration of a genomic disorder. If a syndromic form of schizophrenia is suspected, a referral to a genetics specialist would usually be recommended for diagnostic assessment (58).
What is the clinical relevance of these CNV findings for patients determined to have one of these major CNVs? For 22q11.2 deletions, the relevance is clear: detection significantly changes genetic counseling and anticipatory care from that for other patients with schizophrenia (58). For the 1q21.1 and 15q13.3 deletions, much less is known and there are challenges to providing genetic counseling for individuals with emerging genomic disorders (1, 57). However, detection of these anomalies would be clinically relevant to individuals with these CNVs, their families, and clinicians.
For the patient, genetic counseling would include the 50% risk of transmitting the deletion at each pregnancy, with the caveat that the severity of the phenotype cannot be predicted (58). As for 22q11.2 deletions, prenatal detection would be available (58). Parents of affected individuals should be tested because the deletion may be inherited from a parent. Specific anticipatory care recommendations for 1q21.1 and 15q13.3 deletions await further clinical data and, preferably, larger studies of population comparison subjects. However, history and physical examination, including neurological examination, echocardiogram, and abdominal ultrasound to investigate organ involvement, and routine blood work would also appear warranted (58). For lower-penetrance CNVs, many of which may be inherited from apparently unaffected parents, genetic counseling and clinical recommendations would be even less certain at this time (1, 57). In all cases, a genetics specialist would have the most up-to-date knowledge about these issues.
Neuropsychiatric Perspectives on Phenotype and Implications for Diagnostic Classification
In addition to being directly relevant to clinical practice, these CNV findings challenge some widely held beliefs and point to new research strategies. The data explode a common myth related to the diagnostic specificity of genetic findings. While there has been growing acceptance of the genetic relatedness of schizophrenia and bipolar disorder, there may be some resistance to accepting that expression of an individual CNV may also include autism and other developmental disorders (Figure 2, Table 2). The CNV results suggest a broader neuropsychiatric spectrum of phenotypes. In fact, there is little evidence in medicine for diagnostic purity associated with individual genetic changes. Genetic heterogeneity (many different genetic variants leading to the same phenotype), reduced penetrance (presence of the genetic variant not always expressed as the full disease), and variable expressivity (the same genetic variant leading to various phenotypic expressions) are the norm in human diseases and are apparent with the CNVs associated with schizophrenia. Studies of familial connections between schizophrenia and mental retardation (59, 60) and studies of childhood-onset schizophrenia, a rare clinical subtype in which comorbid autism spectrum disorders are common, also support a neuropsychiatric spectrum involving these developmental conditions (24). On the other hand, the data suggest that although there is some overlap, the pattern of associated CNVs may differ between schizophrenia and autism (Figure 2). For example, while autistic features are commonly reported in children with 22q11.2 deletions who receive psychiatric assessments, 22q11.2 deletions are rarely reported in subjects with autism who are studied (25–27). Also, current data suggest that 1q21.1 duplications are more often present than 1q21.1 deletions in autism (26, 61), whereas the reverse may be the case for schizophrenia (16, 51).
With respect to psychiatric diagnosis, these CNV findings suggest that, as for the dementias, the schizophrenias are beginning to yield to classification based on major causal factors. Comparable to an Alzheimer-type dementia associated with a beta-amyloid precursor protein (APP) gene mutation, a schizophrenia that is related to a 22q11.2 deletion (and perhaps a 1q21.1 or 15q13.3 deletion) may be considered a subtype with distinct management implications.
The findings may also prompt consideration of historical concepts of schizophrenia as an "epiphenomenon" of mental retardation. In fact, schizophrenia is not associated with most forms of mental retardation. The majority of individuals with 22q11.2 deletions, including those with schizophrenia, do not have mental retardation (34, 62), and this will probably also be the case for 1q21.1 and 15q13.3 deletions and lower-penetrance CNVs. Nonetheless, dual-diagnosis populations with schizophrenia and mental retardation are likely to be enriched for these recurrent CNVs, providing a much greater window on the etiology of pfropfschizophrenie than have chromosomal abnormalities detectable by karyotype.
Recurrent De Novo CNVs and a General Mutational Mechanism
Recurrent rare CNVs such as the 22q11.2, 1q21.1, and 15q13.3 deletions may also provide some concrete proof of a long-suspected mutational mechanism in schizophrenia. Over 50 years ago, geneticists Lionel Penrose (63) and Jan Böök (64) proposed that elevated rates of new mutations were likely in schizophrenia. Decreased reproductive fitness in schizophrenia (negative selection) in the face of a steady population prevalence of schizophrenia supports this possibility (65). The mechanism of the recurrence of the associated major CNVs, involving segmental duplications and nonallelic homologous recombination (3, 19), can account for the persistence of these mutations despite the biological disadvantage (reduced fitness) associated with their full expression. It is tantalizing to speculate about accumulating rare CNVs in schizophrenia and CNVs disrupting multiple genes in relevant pathways (14), but these possibilities remain to be proven. Certainly, the relationship of CNVs to reproductive fitness, natural selection, disease prevalence, and human evolution is of intense interest in genetics (9).
Implications for Gene Identification
Multiple independent reports of CNVs overlapping specific genes in patients with schizophrenia may show convergence on individual genes and/or pathways of interest that could assist in understanding pathogenesis. The mechanism of action of CNVs on the expressed phenotype is hypothesized to include effects of the copy number change, e.g., dosage effects, on genes within the CNV and perhaps extending as far as several megabases adjacent ("position" effect). There may be disruption of genes at the CNV break points. Sequence changes, e.g., on the intact chromosome in the case of deletions, may also be a factor. It is important to recognize that copy number gains such as duplications can affect gene expression, and in many cases these effects are similar to the effects of deletions involving the same chromosomal region.
In contrast to the situations for autism and other neuropsychiatric disorders, identifying rare mutations in single genes implicated by multigene CNVs remains a hypothetical possibility for schizophrenia. Could family studies be redesigned to help localize such rare mutations in CNV-related single genes? The phenotypic spectrum associated with the CNVs may provide a clue. One could predict that inherited (familial) forms of "pure" schizophrenia would differ in associated regions from those identified by de novo CNV mutations. The findings of a meta-analysis of linkage studies of familial schizophrenia are consistent with this prediction (66). Rare families with both schizophrenia and autism or mental retardation segregating (59, 60) may represent a better strategy for subtypes of schizophrenia that are associated with CNVs and/or with mutations in key genes affected by such CNVs.
Conclusions
We are in an exciting new era of identifying specific etiologies for individual subtypes of schizophrenia that have important implications for clinical practice in the genomic era. Even though compelling mutations in individual genes have not yet been identified, the campaign to understand the genetic heterogeneity of this complex disorder has begun. Just as for autism, diagnostic classification systems may now begin to delineate genetic subtypes associated with CNVs that represent up to 2% of schizophrenia. Despite significant limitations, the recent genome-wide studies of copy number add to established results for 22q11.2 deletions and point to a more general mutational mechanism involving rare CNVs that elevate risk for schizophrenia, especially more developmental forms of the disease. The findings provide insight into a broader neuropsychiatric spectrum for schizophrenia than previously conceived and indicate new directions for genetic studies. Future studies of CNVs that focus as much on phenotype as on technological advances promise further clinically relevant results and discoveries of genetic pathways to schizophrenia and other psychiatric diseases.
1 : Challenges and standards in integrating surveys of structural variation. Nat Genet 2007; 39(7 suppl):S7–S15 Crossref, Medline, Google Scholar
2 : Whole genome scanning: resolving clinical diagnosis and management amidst complex data. Pediatr Res 2009; 66:357–363 Crossref, Medline, Google Scholar
3 : Genome architecture, rearrangements and genomic disorders. Trends Genet 2002; 18:74–82 Crossref, Medline, Google Scholar
4 : Global variation in copy number in the human genome. Nature 2006; 444:444–454 Crossref, Medline, Google Scholar
5 : Detection of large-scale variation in the human genome. Nat Genet 2004; 36:941–951 Crossref, Google Scholar
6 : Large-scale copy number polymorphism in the human genome. Science 2004; 305:525–528 Crossref, Medline, Google Scholar
7 : Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat 2009; 30:135–144 Crossref, Medline, Google Scholar
8 : Origins and functional impact of copy number variation in the human genome. Nature (in press) Google Scholar
9 : Reduced purifying selection prevails over positive selection in human copy number variant evolution. Genome Res 2008; 18:1711–1723 Crossref, Medline, Google Scholar
10 : Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet 2007; 39(7 suppl):S48–S54 Crossref, Medline, Google Scholar
11 : Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 2008; 40:1166–1174 Crossref, Medline, Google Scholar
12 : Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 1998; 14:417–422 Crossref, Medline, Google Scholar
13 : Schizophrenia and 22q11.2. deletion syndrome. Curr Psychiatry Rep 2008; 10:148–157 Crossref, Medline, Google Scholar
14 : Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008; 320:539–543 Crossref, Medline, Google Scholar
15 : Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 2008; 40:880–885 Crossref, Medline, Google Scholar
16 : Large recurrent microdeletions associated with schizophrenia. Nature 2008; 455:232–236 Crossref, Medline, Google Scholar
17 : Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455:237–241 Crossref, Medline, Google Scholar
18 : New strategies and emerging technologies for massively parallel sequencing: applications in medical research (editorial). Genome Med 2009; 1:40 Crossref, Google Scholar
19 : Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 2009; 84:339–350 Crossref, Medline, Google Scholar
20 : Segmental copy number variation shapes tissue transcriptomes. Nat Genet 2009; 41:424–429 Crossref, Medline, Google Scholar
21 : Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007; 315:848–853 Crossref, Medline, Google Scholar
22 : Structural variation in the human genome. Nat Rev Genet 2006; 7:85–97 Crossref, Medline, Google Scholar
23 : Genetic insights into the neurodevelopmental hypothesis of schizophrenia. Schizophr Bull 2001; 27:417–430 Crossref, Medline, Google Scholar
24 : The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry 2005; 10:434–449 Crossref, Medline, Google Scholar
25 : Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008; 9:341–355 Crossref, Medline, Google Scholar
26 : Copy-number variations associated with neuropsychiatric conditions. Nature 2008; 455:919–923 Crossref, Medline, Google Scholar
27 : Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008; 82:477–488 Crossref, Medline, Google Scholar
28 : Strong association of de novo copy number mutations with autism. Science 2007; 316:445–449 Crossref, Medline, Google Scholar
29 : Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet 2008; 40:1199–1203 Crossref, Medline, Google Scholar
30 : A study of rare structural variants in schizophrenia patients and normal controls from Chinese Han population. Mol Psychiatry 2008; 13:911–913 Crossref, Medline, Google Scholar
31 : Assessment of the frequency of the 22q11 deletion in Afrikaner schizophrenic patients. Am J Med Genet B Neuropsychiatr Genet 2004; 129B:20–22 Crossref, Medline, Google Scholar
32 : Sex chromosome anomalies in childhood onset schizophrenia: an update. Mol Psychiatry 2008; 13:910–911 Crossref, Medline, Google Scholar
33 : Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 2008; 359:1685–1699 Crossref, Medline, Google Scholar
34 : Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum Mol Genet 2008; 17:4045–4053 Crossref, Medline, Google Scholar
35 : The molecular genetics of the 22q11-associated schizophrenia. Brain Res Mol Brain Res 2004; 132:95–104 Crossref, Medline, Google Scholar
36 : Copy-number variation in control population cohorts. Hum Mol Genet 2007; 16:168–173 Crossref, Google Scholar
37 : Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet 2007; 122:345–353 Crossref, Medline, Google Scholar
38 : Chromosome 22q11 deletions are not found in autistic patients identified using strict diagnostic criteria, IMGSAC: International Molecular Genetics Study of Autism Consortium. Am J Med Genet 2000; 96:15–17 Crossref, Medline, Google Scholar
39 : The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry 2006; 45:1104–1113 Crossref, Medline, Google Scholar
40 : Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities (letter). Nat Genet 2008; 40:1466–1471 Crossref, Medline, Google Scholar
41 : A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet 2008; 40:322–328 Crossref, Medline, Google Scholar
42 : Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet 2009; 46:382–388 Crossref, Google Scholar
43 : Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet 2008; 17:458–465 Crossref, Medline, Google Scholar
44 : Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet 2009; 18:988–996 Crossref, Medline, Google Scholar
45 : Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am J Hum Genet 2008; 83:504–510 Crossref, Medline, Google Scholar
46 : CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry 2008; 13:261–266 Crossref, Medline, Google Scholar
47 : A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet 2009; 5(2):e1000373 Crossref, Google Scholar
48 : Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet 2009; Sept 24, Epub ahead of print Google Scholar
49 : Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358:667–675 Crossref, Medline, Google Scholar
50 : Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008; 17:628–638 Crossref, Medline, Google Scholar
51 : Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet 2009; 18:1497–1503 Crossref, Medline, Google Scholar
52 : Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 2009; 84:148–161 Crossref, Medline, Google Scholar
53 : Premature death in adults with 22q11.2. deletion syndrome. J Med Genet 2009; 46:324–330 Crossref, Medline, Google Scholar
54 : 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 2009; 41:160–162 Crossref, Medline, Google Scholar
55 : CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 2003; 82:1–9 Crossref, Medline, Google Scholar
56 : Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 2009; June 23, Epub ahead of print Google Scholar
57 : Emerging microdeletion and microduplication syndromes: the counseling paradigm. Eur J Med Genet 2009; 52:75–76 Crossref, Medline, Google Scholar
58 : Schizophrenia and psychotic disorders, in Psychiatric Genetics: Applications in Clinical Practice. Edited by Smoller JWSheidley BRTsuang MT. Arlington, Va, American Psychiatric Publishing, 2008, pp 99–130 Google Scholar
59 : Elevated rates of schizophrenia in a familial sample with mental illness and intellectual disability. J Intellect Disabil Res 2004; 48:531–539 Crossref, Medline, Google Scholar
60 : 'Pfropfschizophrenie' revisited: schizophrenia in people with mild learning disability. Br J Psychiatry 1998; 173:145–153 Crossref, Medline, Google Scholar
61 : Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007; 39:319–328; correction, 39:1285 Crossref, Medline, Google Scholar
62 : The schizophrenia phenotype in 22q11 deletion syndrome. Am J Psychiatry 2003; 160:1580–1586 Link, Google Scholar
63 : Mutation in man. Acta Genetica 1956; 6:169–182 Medline, Google Scholar
64 : Schizophrenia as a gene mutation. Acta Genetica 1953; 4:133–139 Medline, Google Scholar
65 : Reproductive fitness in familial schizophrenia. Schizophr Res 1996; 21:151–160 Crossref, Medline, Google Scholar
66 : Meta-analysis of 32 genomewide linkage studies of schizophrenia. Mol Psychiatry 2009; 14:774–785 Crossref, Medline, Google Scholar

