
Altered Expression of Genes Involved in GABAergic Transmission and Neuromodulation of Granule Cell Activity in the Cerebellum of Schizophrenia Patients
Abstract
Objective: Deficits in gamma-aminobutyric acid (GABA) signaling have been described in the prefrontal cortex, limbic system, and cerebellum in individuals with schizophrenia. The purpose of the present study was to further investigate cerebellar gene expression alterations as they relate to decreases in GABAergic transmission by examining the expression of GABAergic markers, N -methyl- d -aspartic-acid (NMDA) receptor subunits, and cerebellum neuromodulators in individuals with schizophrenia. Method: Subjects were postmortem men with a diagnosis of schizophrenia (N=13) and a postmortem interval-matched non-psychiatric male comparison group (N=13). The authors utilized real-time-quantitative polymerase chain reaction (PCR) to measure mRNA levels of the following GABAergic markers: glutamic acid decarboxylase (GAD) 65 and 67; GABA plasma membrane transporter-1 (GAT-1); GABA type A (GABA A ) receptor subunits α 6 , β 3 , and δ; and parvalbumin. In addition, real-time-quantitative PCR was utilized to assess mRNA levels of the NMDA receptor (NR) subunits NR1, NR2-A, NR2-B, NR2-C, and NR2-D as well as the cerebellar neuromodulators glutamate receptor (GluR)-6, kainate-preferring glutamate receptor subunit-2 (KA2), metabotropic glutamate receptor (mGluR)-2 and mGluR3, and neuronal nitric oxide synthase. Measurements for mRNA levels were determined using lateral cerebellar hemisphere tissue from both schizophrenia and comparison subjects. Results: Schizophrenia subjects showed significant decreases in mRNA levels of GAD 67 , GAD 65 , GAT-1, mGluR2, and neuronal nitric oxide synthase. Increases in GABA A -α 6 and GABA A -δ as well as GluR6 and KA2 were also observed. Medication effects on the expression of the same genes were examined in rats treated with either haloperidol (Sprague-Dawley rats [N=16]) or clozapine (Long-Evans rats [N=20]). Both haloperidol and clozapine increased the levels of GAD 67 in the cerebellum and altered the expression of other cerebellar mRNAs. Conclusions: These findings suggest that GABA transmission is decreased in the cerebellar cortices in individuals with schizophrenia and additional gene expression changes may reflect an attempt to increase GABA neurotransmission at the cerebellar glomerulus.
One of the most consistent molecular findings from schizophrenia research is the dysfunction of specific subsets of gamma-aminobutyric acid (GABA) expressing interneurons (1 – 4) . Reductions in mRNA and protein levels of glutamic acid decarboxylase 67 (GAD 67 ) have been found in specific interneurons of the prefrontal cortex (5 – 7) and limbic system in individuals with schizophrenia (8 , 9) , and similar findings have been observed in analyses of whole cerebellar tissue (10 , 11) . The same prefrontal cortex interneurons with GAD 67 reductions have also shown decreases in the presynaptic GABA plasma membrane transporter-1 (GAT-1) (12) . Compensatory postsynaptic changes, such as increases in GABA type A (GABA A )-α 2 receptor density and GABA A receptor radioligand binding, in the prefrontal cortex and anterior cingulate cortex have also been reported (13 – 15) . In addition, single nucleotide polymorphisms (SNPs) in the promoter region of the GAD 67 gene expression have been associated with gray matter reductions in patients with childhood onset schizophrenia (16) and decreased cognitive functioning in adult schizophrenia (17) . Given the role of GABAergic interneurons in the modulation of excitatory output, dysfunction in specific interneurons may mediate some of the positive, negative, and cognitive symptoms seen in schizophrenia.
Aside from its role in motor coordination, the cerebellum has been shown to contribute to cognitive functioning (18) through connections of the posterior lateral hemispheres to the prefrontal cortex (19) . Thus, physiological deficits in this brain region could contribute to the pathophysiology of schizophrenia. Supporting this hypothesis, neuroimaging studies have described increased blood flow and glucose consumption in the cerebellum among schizophrenia patients (20 – 22) . Molecular studies have revealed decreased cerebellar expression of the GABAergic protein reelin and the GABA synthesizing enzymes GAD 67 and GAD 65(10 , 11) as well as increased expression of the axonal chemorepellent Semaphorin 3A (23) and increased expression of granule cell-selective, activity-dependent, brain-derived neurotrophic factor, and growth-associated protein of 43 kDa (GAP-43) genes (24) . To further explore the function of cerebellar interneurons in schizophrenia, we investigated changes in transcript levels of GABAergic markers, N -methyl- d -aspartate (NMDA) receptor subunits, and local circuit neuromodulators of GABA release using total RNA prepared from the posterior lateral cerebellar hemispheres of postmortem male schizophrenia subjects and age- and postmortem interval-matched male comparison subjects. Alterations in specific GABAergic transcripts point to a common pathophysiological mechanism across different brain regions and implicate the cerebellum in the cognitive and motor dysfunction observed in individuals with schizophrenia.
Method
Human Subjects
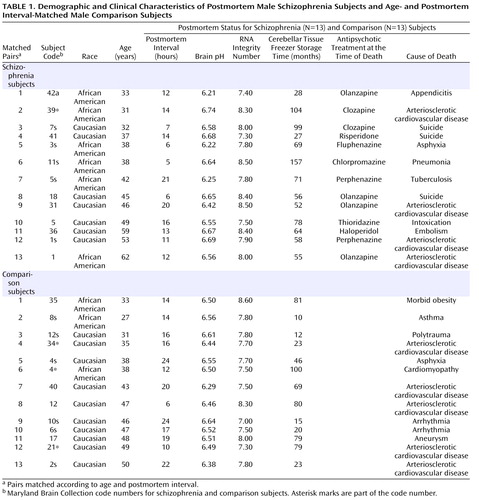
Subjects were postmortem men (N=13) with a diagnosis of schizophrenia (according to DSM-IV criteria) and a sex-, age-, and postmortem interval-matched comparison group (N=13). Cerebellar tissue from both schizophrenia and comparison subjects was acquired from the Maryland Brain Collection ( Table 1 ). All subjects were between 25 and 65 years of age at the time of death, with a postmortem interval ≤24 hours. None of the subjects had a history of alcohol abuse or dependence. No significant differences were found between schizophrenia and comparison subjects for the following factors: 1) mean age (schizophrenia group: 43.5 years [SD=11], range=31–62 years; comparison group: 40.9 years [SD=8], range=27–50 years); 2) mean postmortem interval (schizophrenia group: 12.1 hours [SD=5.1], range=5–21 hours; comparison group: 16.5 hours [SD=5.4], range=6–24 hours); 3) mean brain pH (schizophrenia group: 6.53 [SD=0.2], range=6.21–6.74; comparison group: 6.50 [SD=0.1], range=6.29–6.64); 4) mean RNA integrity number (schizophrenia group: 7.98 [SD=0.12], range=7.30–8.50; comparison group: 7.73 [SD=0.11], range=7.00–8.60); and 5) mean freezer storage time (schizophrenia group: 71 months [SD=24], range=27–157 months; comparison group: 49 months [SD=29], range=10–100 months). At the time of death, six subjects were receiving typical antipsychotics and seven were receiving atypical antipsychotics. Neither schizophrenia nor comparison subjects were receiving medications for mood disorders at the time of death. The subjects’ cerebellar tissue originated from the lateral cerebellar hemisphere corresponding to crus I of lobule VIIa, which has projections to the prefrontal cortex (19) . Specimens were dissected at –20°C and frozen at –80°C until ready for use.
Haloperidol-Treated Rats
The RNA from the cerebellum of Sprague-Dawley rats (N=16) treated with haloperidol was acquired from a previous study (25) . The rats were injected (intramuscular) with either haloperidol depot (38 mg/kg/month [corresponding to approximately 1 mg/kg/day]) or sesame oil (vehicle [comparison group]). After a 7-day acclimation period, the rats were injected once a month for 6 months. They were anesthetized using isoflurane and decapitated 1 month after the last injection. The cerebellum of each rat was removed intact, frozen on dry ice, and stored at –80°C.
Clozapine-Treated Rats
Adult male Long-Evans rats (N=20) were injected (intraperitoneal) with either clozapine (10 mg/kg/day), dissolved in 0.4% acetic acid in 0.9% saline, or a vehicle (comparison group) for 21 days. This paradigm was modified from methods described by Bai et al. (26) , which suggested a maximum D 2 receptor occupancy for clozapine. The cerebellum of each rat in both the clozapine-treated group and vehicle-treated comparison group was removed 18 hours after the last injection, and the lateral hemispheres were dissected and frozen at –80°C.
Quantitative Real-Time Polymerase Chain Reaction (PCR)
All experimental procedures and data analyses were performed blind to the diagnosis. The cerebellar tissue from both human and rat subjects was homogenized using a Polytron homogenizer (Brinkmann Instruments, Westbury, N.Y.). Total RNA was isolated using TriReagent (Sigma, St. Louis) and analyzed with Bioanalyzer 2100 (Agilent Technologies, Santa Clara, Calif.). As detailed in Table 1 , all RNA integrity number values were ≥7.0. The cDNA was synthesized from 4 mg of the total RNA using MMLV Reverse Transcriptase (Promega, Madison, Wisc.). Real-time-quantitative PCRs were run via an Applied Biosystems (Foster City, Calif.) 7000, 7300, or 7500 fast-quantitative real-time PCR machine. Gene expression levels for all human tissue samples as well as those for tissue samples of neuroleptic-treated rats were examined using SYBR Green (Applied Biosystems, Foster City, Calif.), with the exception of the GABAergic markers GABA A -α 6 and GABA A -δ, which were analyzed via TaqMan Assays-on-Demand probes (Applied Biosystems, Foster City, Calif.). All gene expression levels were examined according to the manufacturers’ protocols. Exon spanning primer pairs (Operon, Huntsville, Ala.) specific to 1) GABAergic markers GABA A -β 3 , GAD 67 , GAD 65 , parvalbumin, and GAT-1; 2) NMDA receptor (NR) subunits NR1, NR2-A, NR2-B, NR2-C, and NR2-D; and 3) cerebellar neuromodulators metabotropic glutamate receptor (mGluR)-2 and mGluR3, kainate-preferring glutamate receptors GluR6 and KA2, and neuronal nitric oxide synthase were designed using Primer Express 3.0 (Applied Biosystems, Foster City, Calif. [see the table in the data supplement accompanying the online version of this article]). Expression levels of genes of interest were normalized to those of β-actin, and expression levels were reconfirmed using cyclophilin for selected genes. Validation curves for all primer sets were run with housekeeping genes and shown to have a linear slope of <|0.1|. Dissociation curves for all SYBR Green primer sets showed no evidence of dimerization.
Data and Statistical Analysis
Cerebellar tissue samples from the 13 schizophrenia subjects and 13 comparison subjects were run in triplicate in three separate plates and compared with β-actin or cyclophilin on the same plate. The relative levels of expression of each mRNA were corrected by housekeeping genes according to a previously described method (24) , and the relative GABAergic transcript levels for schizophrenia subjects versus comparison subjects were calculated using the 2 –ΔΔct method (27) . The averaged values per tissue sample were entered into Prism 4.0 (GraphPad Software, San Diego) and analyzed using the Wilcoxon Signed-Rank Test for pair-matched human subjects and Student’s t tests for rats. For further analysis of human subjects, SSPS 15.0 for Windows (SPSS, Chicago) was employed, which included analyses of covariance (ANCOVA) to determine the effect of age, postmortem interval, brain pH, RNA integrity number, and freezer storage time at the levels of each GABAergic transcript. Initial analyses demonstrated no significant effects of brain pH and postmortem interval at the levels of any of the transcripts. Thus, for all subsequent analyses, diagnosis was used as the independent variable, and age, RNA integrity number, and freezer storage time were used as covariates. A bivariate Spearman’s r correlation analysis was employed to examine putative gene-gene interactions. Any statistical analysis with a p value <0.05 was considered significant.
Results
GABAergic Marker Expression in the Lateral Cerebellar Hemisphere of Schizophrenia Subjects
Given the evidence of GABA dysfunction among individuals with schizophrenia, we sought to characterize the condition of inhibitory interneurons in the cerebellum of schizophrenia subjects in order to 1) investigate the mechanisms underlying cerebellar abnormalities and 2) examine the nature and extent of GABA dysfunction in different neuronal populations. Initial studies examined the expression of seven markers of GABA function in the lateral cerebellar hemisphere of 13 pair-matched schizophrenia and comparison subjects using real-time PCR. To normalize the levels of a specific mRNA, β-actin was utilized because the expression of this particular mRNA did not differ between schizophrenia and comparison subjects (see the figure in the data supplement accompanying the online version of this article). Analysis of GABAergic transcript levels in schizophrenia subjects and matched comparison subjects using the Wilcoxon Signed-Rank Test revealed significant decreases in mRNA levels of GAD 67 , GAD 65 , and GAT-1 (schizophrenia to comparison group ratio=0.62, 0.74, and 0.57, respectively). However, no decreases in mRNA levels of parvalbumin were found ( Table 2 , Figure 1 ). Similar decreases in these transcripts were observed when cyclophilin was used as the housekeeping gene (see the data supplement accompanying the online version of this article). Subjects with schizophrenia also showed significant increases in mRNA levels for the granule-cell extrasynaptic GABA A receptor subunits α 6 (schizophrenia to comparison group ratio=1.44) and δ (schizophrenia to comparison group ratio=1.53) relative to β-actin mRNA. However, no changes were found in GABA A -β 3 mRNA, which is expressed in several cell types. In addition to pairwise analyses, we evaluated the effect of potential confounding variables, such as age, freezer storage time, and RNA integrity number, using ANCOVA. As detailed in Table 2 , ANCOVA results confirmed that mRNA levels of GAD 67 , GAT-1, GABA A -α 6, and GABA A -δ were significantly different for schizophrenia subjects relative to comparison subjects (p<0.05), while mRNA levels of GAD 65 were close to significant (p=0.06).
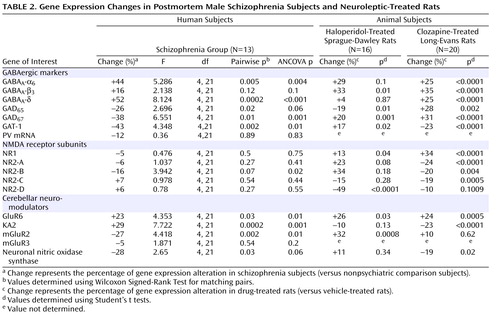
a The top row illustrates significant decreases seen in GABAergic marker transcript expression levels for GAD 67 and GAT-1 and significant increases seen in GABA A -α 6 and GABA A -δ among schizophrenia subjects (Wilcoxon Signed-Rank Test [see Table 2]). Expression levels are relative to β-actin. Values show mRNA levels in matched pairs and averages for schizophrenia and comparison subjects. The bottom row illustrates GABAergic marker transcript expression levels for GAD 67 , GAD 65 , and GAT-1, which are all significantly correlated using Spearman’s r correlation coefficient.
To examine possible gene-gene interactions, mRNA levels were analyzed using Spearman’s r correlations. In the schizophrenia group, positive correlations were 1) GAD 67 with GAD 65 (r=0.922, p<0.001) and 2) GAT-1 (r=0.797, p=0.001) and GAD 65 with GAT-1 (r=0.770, p=0.002) ( Figure 1 ). Comparison subjects also showed a positive correlation between GAD 67 and GAT-1 (r=0.646, p=0.02).
NMDA Receptor Subunit Expression in the Lateral Cerebellar Hemisphere of Schizophrenia Subjects
In addition to GABA deficits, NMDA dysfunction has been found in schizophrenia patients (28) . In the present study, analyses of the levels of NMDA receptor subunits NR1, NR2-A, NR2-B, NR2-C, and NR2-D, using the Wilcoxon Signed-Rank Test, revealed no significant changes except for a tendency toward decrease in the Golgi cell-selective subunit NR2-B (29) (schizophrenia to comparison group ratio=0.84, p=0.07 [ Table 2 ]). Furthermore, when considering the effect of age, freezer storage time, and RNA integrity number using ANCOVA, we found a significant decrease in the levels of NR2-B among schizophrenia subjects (p=0.004 [ Table 2 ]). In addition, the levels of NR2-B in the schizophrenia group were positively correlated with the levels of NR1 (r=0.553, p=0.05), NR2-A (r=0.553, p=0.05), and NR2-D (r=0.663, p=0.01).
Cerebellar Neuromodulator Expression in the Lateral Cerebellar Hemisphere of Schizophrenia Subjects
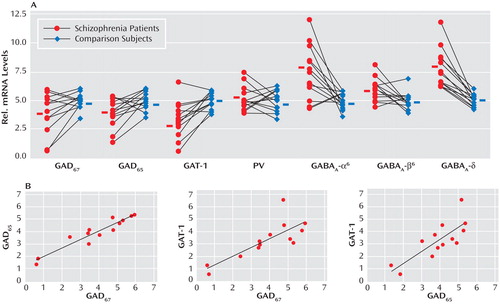
GABA release onto cerebellar granule cells is regulated at the cerebellar glomerulus, where pontine mossy fibers and Golgi cells synapse onto granule-cell dendrites in a close structural and functional unit. Thus, we examined the major modulatory factors of this unit, which could contribute to decreased GABA neurotransmission to granule cells. Of these modulatory factors, we observed significant decreases in GABAergic transcript levels of neuronal nitric oxide synthase (schizophrenia to comparison group ratio=0.72), present in granule cells and molecular layer interneurons (30) , and the Golgi cell-specific mGluR2 (31) (schizophrenia to comparison group ratio=0.73) but not mGluR3 ( Figure 2 ). Interestingly, both nitric oxide production by neuronal nitric oxide synthase and presynaptic mGluR2 receptor activation modulate GABA release from Golgi cells (32) . Moreover, we found significantly increased levels of the glutamatergic kainate receptor subunits GluR6 (schizophrenia to comparison group ratio=1.23) and the granule cell-specific subunit KA2 (schizophrenia to comparison group ratio=1.29 [33] ). ANCOVA revealed that all changes in GABAergic transcript levels were significant (p<0.05) except for changes in neuronal nitric oxide synthase, which were near significant (p=0.06 [ Table 2 ]).
a The top row illustrates significant increases seen in the levels of the kainate receptor subunits GluR6 and KA2 and significant decreases found in levels of mGluR2 and neuronal nitric oxide synthase among schizophrenia subjects (Wilcoxon Signed-Rank Test [see Table 2]). Expression levels are relative to β-actin. Values show mRNA levels in matched pairs and averages for schizophrenia and comparison subjects. The bottom row illustrates the significant correlations between markers expressed in the Golgi-granule cell circuit (using Spearman’s r correlation coefficient).
Correlations of Transcripts Levels in Golgi Cells, Granule Cells, and Local Cerebellar Circuitry Components
To further investigate the functional integrity of the cerebellar glomerulus in schizophrenia subjects, interactions between specific markers in the Golgi-granule cell circuit were analyzed. We found positive correlations for the Golgi cell-selective NR2-B with GAD 67 (r=0.922, p<0.001), GAD 65 (r=0.854, p<0.001), and GAT-1 (r=0.671, p=0.01) and for mGluR2 with GAD 65 (r=0.575, p=0.04), all of which were decreased in the schizophrenia group. In addition, GABAergic transcripts expressed in granule cells showed correlations for NR2-C with GABA A -α 6 (r=0.639, p=0.02), GABA A -β 3 (r=0.722, p=0.005), GABA A -δ (r=0.652, p=0.02), and KA2 (r=0.726, p=0.005), which were all increased among schizophrenia subjects. Finally, we found that neuronal nitric oxide synthase correlated with GABA A -β 3 (r=0.757, p=0.003); GABA A -α 6 correlated with GluR6 (r=0.659, p=0.01); and KA2 correlated with GABA A -δ (r=0.600, p=0.03).
Positive correlations for components of Golgi cell-granule cell circuitry were 1) NR2-B with GABA A -β 3 (r=0.600, p=0.03), GluR6 (r=0.594, p=0.03), and neuronal nitric oxide synthase (r=0.624, p=0.02) and 2) mGluR2 with GluR6 (r=0.713, p=0.006) and GABA A -α 6 (r=0.797, p<0.001). Significant negative correlations were observed for GAT-1 with NR2-C (r=–0.553, p=0.05) and KA2 (r=–0.779, p=0.002), presumably reflecting a compensatory mechanism to increase GABA neurotransmission in the synaptic cleft.
Medication Effects on Gene Expression in Haloperidol- and Clozapine-Treated Rats
Both schizophrenia and comparison subjects were receiving antipsychotics at the time of death, with approximately one-half of the schizophrenia group being treated with typical antipsychotics and one-half being treated with atypical antipsychotics ( Table 1 ). Analysis of gene expression levels between schizophrenia subjects who were receiving atypical versus typical antipsychotics showed a significant increase in GAD 67 gene expression for the former (p=0.03 [Mann-Whitney Rank Sum Test]). This finding suggests that medication type may have an effect on gene expression levels in individuals with schizophrenia. Therefore, we analyzed the levels of the same mRNAs in the cerebellum of rats chronically treated with approximately 1 mg/kg/day of the typical neuroleptic haloperidol. As detailed in Table 2 , we found significant increases in GABA A -β 3 , GAD 67 , GAT-1, NR1, GluR6, and mGluR2 in these rats. However, a significant decrease was seen in the expression of GAD 65 and NR2-D transcripts.
To examine the effects of atypical antipsychotics, rats were treated with clozapine for 21 days (10 mg/kg/day). As detailed in Table 2 , these animals expressed increased mRNA levels of GABA A -β 3 , GAD 67 , and GAD 65 , while GAT-1 expression was significantly decreased. The NMDA receptor subunit NR1 was significantly increased, whereas NR2-A, NR2-B, and NR2-C were all decreased. Finally, GluR6 was significantly increased, KA2 and neuronal nitric oxide synthase were decreased by clozapine treatment. Overall, our results indicate that clozapine could compensate, in part, for the decreased level of GABA synthesizing enzymes and other gene expression changes observed in schizophrenia subjects.
Discussion
An increasing number of studies support the hypothesis that GABA neurotransmission is deficient in individuals with schizophrenia (2 – 4) . Although GABAergic deficits have been well characterized in the prefrontal cortex (5 – 7) and limbic system (8 , 9) , few studies have examined these deficits in the cerebellum (10 , 11) . The present study demonstrated GABA transmission deficits in the lateral cerebellar hemisphere in individuals with schizophrenia, which implicates selective impairment in local granule-cell Golgi-cell communication, along with altered expression of neuromodulators of granule cell activity. Disinhibition of granule cells by deficient tonic inhibition from Golgi cells could disrupt cerebellar output by Purkinje cells, which in turn may affect other brain regions in the cortico-cerebellar-thalamic-cortical circuit, including the prefrontal cortex (34) .
Several discrete GABAergic interneurons are located in the cerebellum, most notably Golgi cells in the granule layer and basket and stellate cells in the molecular layer. Although basket and stellate cells provide feed-forward inhibition from granule cells to Purkinje cells, Golgi cells provide the sole source of feedback inhibition to granule cells (35) . All of these interneurons express the GABA synthesizing enzymes GAD 65 and GAD 67 . Our findings indicate that the levels of these GABAergic transcripts are decreased in individuals with schizophrenia relative to comparison subjects, suggesting that GABA synthesis may be impaired in schizophrenia patients. Consistent with our findings, Fatemi et al. (11) reported decreases in protein levels of both GAD 65 and GAD 67 in the cerebellum of a separate cohort of schizophrenia subjects.
Although GABA synthesis cannot be directly measured in postmortem tissue, other markers can be investigated to infer whether GABAergic transmission is reduced. For example, decreased levels of the presynaptic GAT-1 may reflect a compensatory attempt to increase synaptic GABA. Mature Golgi and basket/stellate cells express GAT-1, while Purkinje cells and glia do not (36) , suggesting that GAT-1 deficits may be restricted to specific GABAergic interneurons. This is consistent with a similar decrease in GAT-1 observed in the prefrontal cortex of schizophrenia patients in association with GAD 67 deficits (12) . Interestingly, the calcium binding protein parvalbumin, which has been found to be decreased in the prefrontal cortex in individuals with schizophrenia (6) , was not decreased in the present study, suggesting that different subtypes of interneurons may be affected in the cerebellum. Parvalbumin is expressed primarily in Purkinje and basket/stellate cells but only in a small subpopulation of Golgi cells (37) . Conversely, the significant increases in the expression of the extrasynaptic GABA A receptor subunits α 6 and d suggest that a compensatory mechanism may occur in order to increase GABA receptivity by granule cells. As a result of freezing the tissue samples, we were unable to confirm the localization of specific transcripts by in situ hybridization. However, our findings point to Golgi cells as the predominately affected cerebellar interneuron in schizophrenia.
A competing, but not mutually exclusive, hypothesis for schizophrenia proposes that NMDA receptor-mediated neurotransmission may be deficient in individuals with schizophrenia (28) and that GABAergic interneurons are particularly vulnerable to NMDA receptor hypofunction (38) . In the present study, GABAergic interneuron function in the cerebellum was further investigated by measuring the expression of cell-selective NMDA receptor subunits. Although each cell type expresses the obligatory NR1 subunit, Purkinje cells express primarily NR2-A; Golgi cells express primarily NR2-B; granule cells express primarily NR2-C; and basket/stellate cells express primarily NR2-D (29) . Among these subunits, we found a decrease in the levels of the Golgi cell-selective NR2-B. The strong correlations between NR2-B expression and GAD 67 , GAD 65 , and GAT-1 expression in the schizophrenia group further suggests that GABA transmission deficits may be localized to Golgi cells. Alternatively, it is possible that the number of Golgi cells and not their activity may be decreased in individuals with schizophrenia.
GABA release by Golgi cells is regulated by various factors in both Golgi and granule cells, including Golgi cell-specific mGluR2 (31) and the granule cell-located neuronal nitric oxide synthase (32) . Activation of both systems normally decreases GABA release by Golgi cells, and both were decreased in our cohort of schizophrenia subjects ( Figure 2 ), presumably compensating for decreased levels of GABA in the synapse. On the other hand, mGluR3, which is expressed in Golgi cells and glia (31) , was not altered in the schizophrenia group. In addition, although decreased levels of mRNA in the receptor subunits GluR6 and KA2 have been previously reported in the prefrontal cortex and hippocampus of schizophrenia patients (33 , 39) , we observed increased cerebellar expression of these kainate receptors. Since both markers are expressed in granule cells and these cells have shown increased expression of activity dependent-genes in the same group of schizophrenia subjects (24) , it is possible that the changes in kainate receptor subunits may reflect a change in basal granule cell activity.
The changes in gene expression and putative cerebellar microcircuit alterations are illustrated in Figure 3 . Although postmortem human tissue studies only provide a limited view of the premortem state and the use of tissue homogenates does not allow localization of transcripts to specific cell types, analyses of gene expression patterns using gene-gene correlations are valuable tools to infer the functional relevance of the observed changes across different cells in the network. An alteration in GABA neurotransmission between Golgi and granule cells is further evidenced by the positive correlations between the modulators of GABA release neuronal nitric oxide synthase and mGluR2 and between the Golgi specific marker NR2-B and neuronal nitric oxide synthase. Moreover, the negative correlations for GAT-1 with the granule cell-specific receptor subunits NR2-C and KA2 suggest a defect in modulation of GABA release by Golgi cells and increased granule cell firing. Although these findings are intriguing, animal models will need to be developed in order to confirm the dynamic nature of the interactions between granule cell and Golgi cell gene-expression patterns reported in the present study.
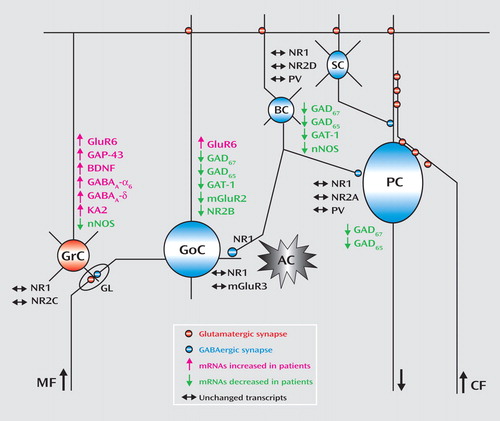
a The graph illustrates decreased GABA gene expression in Golgi cells and concomitant increases in granule-cell gene expression. Expression patterns are based on the Allen Brain Atlas (www.brain-map.org) and data from the literature. PV=parvalbumin; GrC=granule cells; GoC=Golgi cells; BC=basket cells; SC=stellate cells; PC=Purkinje cells; GL=cerebellar glomerulus; AC=astrocyte; MF=mossy fibers (from pontine nuclei); and CF=climbing fibers (from inferior olive).
Our gene expression findings in schizophrenia subjects point to dysfunctional regulation of GABA release between Golgi and granule cells but do not take into account any effects of antipsychotic medication. Although antipsychotics have been shown to affect gene expression in several brain regions, these alterations have not been demonstrated in the cerebellum. As detailed in Table 2 , haloperidol treatment resulted in increased levels of GAD 67 , mGluR2, NR2-B, GAT-1, and neuronal nitric oxide synthase in chronically treated rats, whereas clozapine was more effective in increasing the expression of GAD 67 and GAD 65 . Comparison of the levels of GluR6, GABA A -α 6, and GABA A -δ receptor mRNAs in schizophrenia subjects and treated animals suggests that the observed increases of these transcripts in the schizophrenia group could be explained by medication effects. In contrast, the increases in KA2 mRNA seen in the schizophrenia group could not be attributable to antipsychotic treatment, since both haloperidol and clozapine decreased the levels of KA2 mRNA. In addition, medication effects could account for an underrepresentation of the changes in NR2-C and NR2-D gene expression in the schizophrenia group. Based on the results of the chronic neuroleptic exposure in rats, haloperidol seems to have a more beneficial effect on gene expression in the local cerebellar circuit relative to clozapine. Furthermore, although both medications were effective in increasing GAD 67 expression in rats, neither treatment was effective in schizophrenia subjects. Our results support the idea that combining different types of antipsychotics with GABA A receptor agonists may provide enhanced benefits for treating the underlying GABA-glutamate pathophysiology of schizophrenia.
In summary, we found that the expression of multiple GABAergic markers is decreased in the lateral cerebellar hemisphere among individuals with schizophrenia and this deficit is most likely localized to Golgi cells. Overall, patient mRNA expression data suggest that GABA neurotransmission deficits could be responsible for increased granule cell activity and disinhibition of glutamatergic signaling in the cerebellum, leading to aberrant cerebellar output. These changes may underlie not only the alterations in cerebellar blood flow observed in our schizophrenia cohort, but also, and most importantly, the clinical symptoms associated with abnormal cerebellar function in schizophrenia (40) .
1. Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB: Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry 2005; 57:252–260Google Scholar
2. Akbarian S, Huang HS: Molecular and cellular mechanisms of altered GAD 1 /GAD 67 expression in schizophrenia and related disorders. Brain Res Rev 2006; 52:293–304 Google Scholar
3. Lewis DA, Hashimoto T, Volk DW: Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6:312–324Google Scholar
4. Benes FM, Berretta S: GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001; 25:1–27Google Scholar
5. Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA: Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 2000; 57:237–245Google Scholar
6. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA: Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 2003; 23:6315–6326Google Scholar
7. Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE Jr, Jones EG: Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 1995; 52:258–266Google Scholar
8. Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM: Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 2002; 59:521–529Google Scholar
9. Woo TU, Walsh JP, Benes FM: Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl- d -aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 2004; 61:649–657 Google Scholar
10. Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E: Decrease in reelin and glutamic acid decarboxylase 67 (GAD 67 ) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 2000; 57:1061–1069 Google Scholar
11. Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E: GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and reelin proteins in cerebellum. Schizophr Res 2005; 72:109–122Google Scholar
12. Volk D, Austin M, Pierri J, Sampson A, Lewis D: GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 2001; 158:256–265Google Scholar
13. Benes FM, Vincent SL, Alsterberg G, Bird ED, SanGiovanni JP: Increased GABA A receptor binding in superficial layers of cingulate cortex in schizophrenics. J Neurosci 1992; 12:924–929 Google Scholar
14. Benes FM, Vincent SL, Marie A, Khan Y: Up-regulation of GABA A receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 1996; 75:1021–1031 Google Scholar
15. Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA: Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex 2002; 12:1063–1070Google Scholar
16. Addington AM, Gornick M, Duckworth J, Sporn A, Gogtay N, Bobb A, Greenstein D, Lenane M, Gochman P, Baker N, Balkissoon R, Vakkalanka RK, Weinberger DR, Rapoport JL, Straub RE: GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD 67 ), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol Psychiatry 2005; 10:581–588 Google Scholar
17. Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, Vakkalanka RK, Kolachana BS, Kleinman JE, Weinberger DR: Allelic variation in GAD1 (GAD 67 ) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry 2007; 12:854–869 Google Scholar
18. Schmahmann JD, Sherman JC: The cerebellar cognitive affective syndrome. Brain 1998; 121(pt 4):561–579Google Scholar
19. Ramnani N: The primate cortico-cerebellar system: anatomy and function. Nat Rev Neurosci 2006; 7:511–522Google Scholar
20. Kim JJ, Mohamed S, Andreasen NC, O’Leary DS, Watkins GL, Boles Ponto LL, Hichwa RD: Regional neural dysfunctions in chronic schizophrenia studied with positron emission tomography. Am J Psychiatry 2000; 157:542–548Google Scholar
21. Potkin SG, Alva G, Fleming K, Anand R, Keator D, Carreon D, Doo M, Jin Y, Wu JC, Fallon JH: A PET study of the pathophysiology of negative symptoms in schizophrenia. Am J Psychiatry 2002; 159:227–237Google Scholar
22. Malaspina D, Harkavy-Friedman J, Corcoran C, Mujica-Parodi L, Printz D, Gorman JM, Van Heertum R: Resting neural activity distinguishes subgroups of schizophrenia patients. Biol Psychiatry 2004; 56:931–937Google Scholar
23. Eastwood SL, Law AJ, Everall IP, Harrison PJ: The axonal chemorepellant Semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol Psychiatry 2003; 8:148–155Google Scholar
24. Paz RD, Andreasen NC, Daoud SZ, Conley R, Roberts R, Bustillo J, Perrone-Bizzozero NI: Increased expression of activity-dependent genes in cerebellar glutamatergic neurons of patients with schizophrenia. Am J Psychiatry 2006; 163:1829–1831Google Scholar
25. Bustillo J, Barrow R, Paz R, Tang J, Seraji-Bozorgzad N, Moore GJ, Bolognani F, Lauriello J, Perrone-Bizzozero N, Galloway MP: Long-term treatment of rats with haloperidol: lack of an effect on brain N-acetyl aspartate levels. Neuropsychopharmacology 2006; 31:751–756Google Scholar
26. Bai O, Zhang H, Li XM: Antipsychotic drugs clozapine and olanzapine upregulate bcl-2 mRNA and protein in rat frontal cortex and hippocampus. Brain Res 2004; 1010:81–86Google Scholar
27. Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(–Delta Delta C[T]) method. Methods 2001; 25:402–408Google Scholar
28. Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE Jr, Jones EG: Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci 1996; 16:19–30Google Scholar
29. Scherzer CR, Landwehrmeyer GB, Kerner JA, Standaert DG, Hollingsworth ZR, Daggett LP, Velicelebi G, Penney JB Jr, Young AB: Cellular distribution of NMDA glutamate receptor subunit mRNAs in the human cerebellum. Neurobiol Dis 1997; 4:35–46Google Scholar
30. Baader SL, Schilling K: Glutamate receptors mediate dynamic regulation of nitric oxide synthase expression in cerebellar granule cells. J Neurosci 1996; 16:1440–1449Google Scholar
31. Berthele A, Platzer S, Laurie DJ, Weis S, Sommer B, Zieglgansberger W, Conrad B, Tolle TR: Expression of metabotropic glutamate receptor subtype mRNA (mGluR1-8) in human cerebellum. Neuroreport 1999; 10:3861–3867Google Scholar
32. Wall MJ: Endogenous nitric oxide modulates GABAergic transmission to granule cells in adult rat cerebellum. Eur J Neurosci 2003; 18:869–878Google Scholar
33. Porter RH, Eastwood SL, Harrison PJ: Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res 1997; 751:217–231Google Scholar
34. Andreasen NC, Nopoulos P, O’Leary DS, Miller DD, Wassink T, Flaum M: Defining the phenotype of schizophrenia: cognitive dysmetria and its neural mechanisms. Biol Psychiatry 1999; 46:908–920Google Scholar
35. Ito M: Cerebellar circuitry as a neuronal machine. Prog Neurobiol 2006; 78:272–303Google Scholar
36. Takayama C, Inoue Y: Developmental expression of GABA transporter-1 and 3 during formation of the GABAergic synapses in the mouse cerebellar cortex. Brain Res Dev Brain Res 2005; 158:41–49Google Scholar
37. Bastianelli E: Distribution of calcium-binding proteins in the cerebellum. Cerebellum 2003; 2:242–262Google Scholar
38. Homayoun H, Moghaddam B: NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 2007; 27:11496–11500Google Scholar
39. Meador-Woodruff JH, Davis KL, Haroutunian V: Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology 2001; 24:545–552Google Scholar
40. Ho BC, Mola C, Andreasen NC: Cerebellar dysfunction in neuroleptic naive schizophrenia patients: clinical, cognitive, and neuroanatomic correlates of cerebellar neurologic signs. Biol Psychiatry 2004; 55:1146–1153Google Scholar

