
Informed Consent: Assessment of Comprehension
Abstract
Objective:The authors designed and evaluated a structured and rigorous informed consent procedure involving subjects with schizophrenia.Method:Informed consent forms were read and explained to 49 schizophrenic patients participating in ongoing clinical treatment research trials. The subjects answered a questionnaire relating to each research protocol. Protocol procedures were reiterated until the patients answered 100% of the questions correctly. Subjects were asked the same questions 7 days later to ascertain how much of the information they had retained. Results:The patients’ median score on the first trial of the informed consent questionnaire was 80% correct. To achieve 100% correct responses, 53% of the patients required a second trial of the questionnaire, and 37% of them required three or more trials. Scores improved between the first trial and the trial on day 7. Ninety-six percent of the subjects felt adequately informed, 66% reported participating in the research protocol for personal reasons, and 34% reported participating at the suggestion of others.Conclusions:These findings demonstrate that when adequate informed consent procedures are established, schizophrenic research subjects are able to understand and retain critical components of informed consent information. Am J Psychiatry 1998; 155: 1508-1511
Obtaining informed consent from psychiatric populations has gained substantial attention from researchers aiming to determine patients’ capacity to understand the risks and benefits of participation in clinical research trials (1–4). Empirical research has demonstrated that patients are able to understand and use only a portion of the information provided by consent forms (5–7); this is especially true for patients with the more debilitating mental illnesses such as schizophrenia (8, 9). Previous research has also shown that while schizophrenic research subjects indicated that they understood informed consent material, objective assessment of their understanding sometimes proved otherwise (7). The degree of impairment was related to thought disturbance, but this did not affect acceptance or refusal of antipsychotic medication.
Historically, studies have used various methods for assessing patients’ capacities to comprehend consent form information, mainly through brief yes/no self-report questionnaires. More recently, researchers have raised concerns regarding the adequacy of such assessment and have stressed the necessity of standardizing these procedures (1, 10). Previously published assessments may have been inadequate because legal standards have been vague and inconsistent. In an effort to standardize this procedure, the following “legal standards” for assessing patients’ understanding of consent information have been presented (1): one must have the ability 1) to express a choice, 2) to understand information relevant to the decision about treatment, 3) to appreciate the significance, for one"s own situation, of the information disclosed about the illness and the possible treatments, and 4) to manipulate the information rationally (or reason about it) in a manner that allows one to make comparisons and weigh outcomes. These standards are generally accepted in the legal arena for use in determining patients’ decision-making capacities. They have not been formally applied to determining the ability of schizophrenic subjects to provide consent for research protocols.
The process of giving informed consent can be divided into a phase or segment in which information is transmitted to the prospective subject—who must be able to comprehend the risks, benefits, and significance of participating in the research—and a phase for decision making in which consent is given. In this latter phase, it is presumed, but not empirically documented, that the prospective subject has grasped and digested the relevant information for making a rational decision regarding his or her participation in the research.
The aim of the current study was to design a rigorous informed consent procedure that would not only maximize psychotic patients’ potential for understanding and retaining informed consent material but also adequately assess their ability to do so. Toward this end, we developed an assessment questionnaire that incorporated the standards we have mentioned.
METHOD
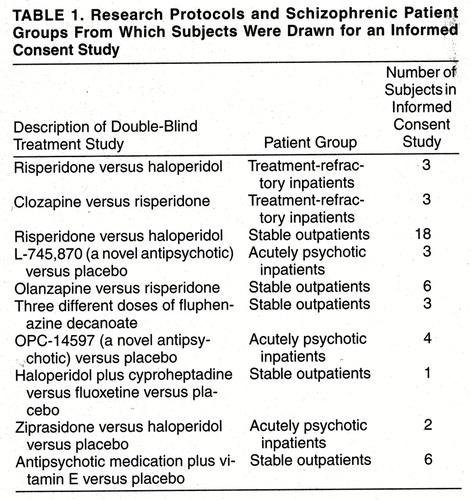
Our data were collected retrospectively from patients included in 10 double-blind clinical research protocols conducted at the West Los Angeles Veterans Affairs (VA) Medical Center (table 1). Each protocol and its informed consent form was approved by the Institutional Review Board of the center. All subjects included in this report gave written informed consent to participate in one of these studies. Moreover, the informed consent procedure described in this report was approved as mandated by the Institutional Review Board and by the Data and Statistical Monitoring Board at the center (which monitors the progress of schizophrenic, Alzheimer’s disease, and other high-risk patients in research studies) for all schizophrenic subjects participating in our research program.
Schizophrenic patients at the West Los Angeles VA Medical Center were approached by their treating physicians regarding their interest in participating in a randomized clinical trial. In cases where the patient was interested, the attending physician described the protocol in some detail. If the patient expressed further interest in being a participant, the study coordinator then thoroughly read the informed consent form to the participant, frequently pausing to assess the level of understanding and answer necessary questions. Upon completion of this procedure, a questionnaire assessing the patient"s comprehension of the informed consent material was administered, and responses were recorded.
This questionnaire, the Informed Consent Survey (appendix 1), was designed by us and incorporates the previously discussed four “legal standards” for assessing patients’ capacities to understand informed consent. For each clinical study, we designed a unique Informed Consent Survey. Items assess patients’ knowledge of the following: specific details and overall goals of the study, patients’ choices as participants in the study, doctors’ responsibilities to the study, and the potential risks of antipsychotic medications. Also, the Informed Consent Survey inquires about the subject’s understanding of his or her physician’s dual role as both clinician and researcher. In cases where the patient did not respond correctly to an item of the questionnaire, that portion of the consent form was re-explained to him or her, and the Informed Consent Survey was then readministered. This procedure was repeated until the patient answered all items of the survey correctly, at which point the informed consent form was signed. The Informed Consent Survey was readministered 7 days later. If a patient failed to understand a critical item at this 1-week postconsent quiz, the item was explained until the patient reported that he or she grasped the concept and answered the survey item correctly. If a patient could not answer the critical questions correctly, he or she was excluded from participation in the research protocol. All subjects were administered the Brief Psychiatric Rating Scale (BPRS) at the completion of the first administration of the Informed Consent Survey. Statistical analyses of the data were performed with SAS software (11).
The 49 subjects were inpatients and outpatients, often in transition from one phase to another. Their mean age was 46.5 years (SD=8.5), and their mean age at onset of illness was 26.5 years (SD=6.4). The majority of the subjects were single (72%, versus 9% married and 19% divorced), were male (94%, versus 6% female), and were veterans (98%, versus 2% nonveterans). The subjects were predominately Caucasian (47%) and African American (39%), with smaller portions being Latino (8%) or Asian (6%). The mean level of education was 13.0 years (SD=1.8). All subjects were free of conservatorship or guardianship and were legally competent to give consent to participate in treatment research; i.e., none of the subjects had had a judicial process to determine competency.
RESULTS
The patients’ median score on the first trial was 80% correct. Fifty-three percent (N=26) of the patients required a second trial to obtain 100% correct, and 37% (N=18) required three or more trials. For the entire study group, scores improved between the first trial and the day 7 trial (McNemar χ2=9.8, df=1, p=0.02). There was a relationship between conceptual disorganization as rated on the BPRS and the percentage of correct responses on the day 7 postconsent trial of the Informed Consent Survey (table 2). Open-ended questions regarding participation in the research were answered as follows: 65% (N=32) of the subjects claimed that they were participating for personal benefits or altruistic reasons, 35% (N=17) claimed that they were participating at the suggestion of others, and 96% (N=47) felt that they were adequately informed.
DISCUSSION
This retrospective study demonstrated that through the implementation of systematic and thorough informed consent procedures, many psychotic patients—but not necessarily all—are able to comprehend and retain critical components of informed consent. Our results may reflect some preselection bias, since we did not approach patients who were so ill that they could not possibly give consent. A majority of the patients required two or more reiterations of the questionnaire in order to demonstrate complete mastery of the material.
We also found that conceptual disorganization was correlated with impaired performance on the Informed Consent Survey when it was readministered 7 days after consent had been given, supporting the findings of a previous study of informed consent in schizophrenia (7). It is notable that even high scores on other psychosis items of the BPRS (i.e., unusual thought content, hallucinations, suspiciousness) did not appear to impair subjects’ learning and retaining key information on the procedures, risks, and benefits of the research protocol (table 2). These findings support previous studies which reported that psychiatric patients, when exposed to the informed consent process on just one occasion, are able to retain only a portion of the informed consent material (5, 7), as well as studies which demonstrated that informed consent procedures that incorporate patient education result in increased comprehension and retention of information (3, 8, 12). Further, these findings suggest that psychiatric symptoms such as hallucinations and delusions do not interfere with competency. Our findings also suggest that there are large differences in the ability of patients with schizophrenia to understand the important elements of consent.
On the basis of these findings, we plan to further evaluate patients’ ability to retain informed consent information over longer periods of time and to correlate their ability to comprehend this information with concurrent assessments of their neurocognitive functioning, particularly, objective measures of verbal memory and abstract reasoning. In addition, we will design and validate training methods that result in enhanced acquisition and durability of the information that is required for genuine informed consent by patients participating in trials of treatments for schizophrenia.
Appendix 1. Informed Consent Survey
Questions (and Their Correct Answers)
| 1. | Do you know what illness is studied at this clinic? (Mental illness or schizophrenia) | ||||
| 2. | If you don’t want to, do you have to be in this study? (No, this is a voluntary study.) | ||||
| 3. | If you don’t want to be in this study, will you still be able to get treatment for your illness somewhere else? (Yes) | ||||
| 4. | If you do not want to be in this study, will you lose any of your disability benefits like your VA benefits or SSI checks? (No) | ||||
| 5. | If you want to, when could you quit this study? (At any time) | ||||
| 6. | True or false?: Your treating doctor is also the investigator of this study. (True) | ||||
| 7. | How long will this study last? (2 years) | ||||
| 8. | In the very beginning of this study, everyone will be taking the same medication for at least 2 weeks. What medication is that? (Haldol[haloperidol]) | ||||
| 9. | What are the two different medications that are being tested in this study? (Haldol and risperidone) | ||||
| 10. | How is it decided who will receive Haldol or risperidone? (A computer randomly decides who will receive risperidone or Haldol.) | ||||
| 11. | True or false?: Half the patients in the study will receive Haldol and half will receive risperidone. (True) | ||||
| 12. | Will you know which medication you are taking? Will the doctor? (No, neither the doctor nor I will know which medication I will be receiving.) | ||||
| 13. | True or false?: There will be a 2-week period of time in the beginning of the study when the amount of medication you take will be decided by the study, and your doctor will not be able to change it. (True) | ||||
| 14. | Please name a serious side effect that a few people have been known to get while taking Haldol and/or risperidone. (One answer needed—tardive dyskinesia or neuroleptic malignant syndrome) | ||||
| 15. | Please name at least two mild side effects that some people may have if they are taking Haldol and/or risperidone. (Patient must name any two side effects.) | ||||
| 16. | If you are a woman, will you have to use an acceptable form of birth control and have pregnancy tests while you are in the study? (Yes) | ||||
| 17. | Is it possible that your symptoms might get better while you are in the study? Is it possible that your symptoms might get worse while you are in the study? (Yes to both parts) | ||||
| 18. | Is it possible that your side effects might get worse? Could they get better? (Yes to both parts) | ||||
| 19. | Please describe two kinds of tests or “assessments” that you may be asked to do during the study. (Memory and/or coordination tests using a computer, blood tests, movement tests, meeting with doctor to discuss symptoms, problem-solving tests that involve the use of a video camera) | ||||
| 20. | Will you be asked to make extra trips to the VA to complete special testing? (Yes) | ||||
| 21. | Will blood be taken throughout the study? (Yes) | ||||
| 22. | How many days per week will you attend group? (Two) | ||||
| 23. | Who in the study is allowed to attend the skills groups? (Everyone in the study will be able to participate in skills training.) | ||||
| 24. | Is there a chance that these groups might be stressful for some people? (Yes) | ||||
| 25. | There is a part of the study that is called IVAST. Can you describe what happens in this program? (It is a program which helps people from the group practice living skills out in the community.) | ||||
| 26. | What are your chances of receiving IVAST? (50%) | ||||
| 27. | If you have any questions about this study, whom can you ask? Where can you find the phone numbers if you wanted to call someone about your questions? (Drs. Marder and Liberman, Doreen Ross, Alix Strough, and other staff at the clinic are available. Phone numbers and locations may be found on the consent form or on the orange card.) | ||||
| 28. | Will this program cost you money, or will you have to pay for anything? (No) | ||||
| 29. | How much money will you receive for travel expenses? ($5.00) | ||||
| 30. | What are some improvements that you could possibly get while in the MRRS II study? (Reduction of my symptoms, better social relationships, reduced chance of having a relapse, and increased quality of life) | ||||
| 31. | After you have finished all groups, then what happens? (I will come in about once a month for follow-up studies until the end of the 2 years.) | ||||
Open-Ended Questions
| 1. | What are some of the reasons that the doctors are doing this study? | ||||
| 2. | What made you decide to become part of this study? | ||||
| 3. | Do you think that this study has been explained to you clearly? | ||||
| 4. | Do you think that you (by yourself) can make the decision to be part of this study? | ||||
| 5. | How would you feel about a rule that said you had to have someone else consent for you? | ||||
Presented as a poster session at the 149th annual meeting of the American Psychiatric Association, New York, May 4–9, 1996Received May 7, 1997; ; revision received March 23, 1998; accepted April 9, 1998. From the Department of Psychiatry, West Los Angeles VA Medical Center; and the Department of Psychiatry and Biobehavioral Science, School of Medicine, University of California at Los Angeles. Address reprint requests to Dr. Donna A. Wirshing, West Los Angeles VA Medical Center, 11301 Wilshire Blvd., Bldg. 210, Room 15, Los Angeles, CA 90073.
 |
 |
1. Grisso T, Appelbaum PS: Comparison of standards for assessing patients’ capacities to make treatment decisions. Am J Psychiatry 1995; 152:1033–1037Link, Google Scholar
2. Grisso T, Appelbaum PS: The MacArthur treatment competence study, III: abilities of patients to consent to psychiatric and medical treatments. Law and Human Behavior 1995; 19:149–174Crossref, Medline, Google Scholar
3. Delano SJ, Hons BA, Zucker JL: Protecting mental health research subjects without prohibiting progress. Hosp Community Psychiatry 1994; 45:601–603Abstract, Google Scholar
4. National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research: The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research: DHEW publication (OS) 78-0013, Appendix II; DHEW publication (OS) 78-0014. Rockville, Md, Department of Health, Education, and Welfare, 1979Google Scholar
5. Grossman L, Summers F: A study of the capacity of schizophrenic patients to give informed consent. Hosp Community Psychiatry 1980; 31:205–206Abstract, Google Scholar
6. Munetz MR, Roth LH: Informing patients about tardive dyskinesia. Arch Gen Psychiatry 1985; 42:866–871Crossref, Medline, Google Scholar
7. Irwin M, Lovitz A, Marder SR, Mintz J, Winslade WJ, Van Putten T, Mills MJ: Psychotic patients’ understanding of informed consent. Am J Psychiatry 1985; 142:1351–1354Link, Google Scholar
8. Kleinman I, Schachter D, Jeffries J, Goldhamer P: Effectiveness of two methods for informing schizophrenic patients about neuroleptic medication. Hosp Community Psychiatry 1993; 44:1189–1191Abstract, Google Scholar
9. Schachter D, Kleinman I, Prendergast P, Remington G, Schertzer S: The effect of psychopathology on the ability of schizophrenic patients to give informed consent. J Nerv Ment Dis 1994; 182:360–362Crossref, Medline, Google Scholar
10. Appelbaum PS: The right to refuse treatment with antipsychotic medications: retrospect and prospect. Am J Psychiatry 1988; 145:413–419Link, Google Scholar
11. SAS ProcGLM (Procedure General Linear Model). Cary, NC, SAS Institute, 1992Google Scholar
12. Brown CS, Wright RG, Christensen DB: Association between type of medication instruction and patients’ knowledge, side effects, and compliance. Hosp Community Psychiatry 1987; 38:55–60Abstract, Google Scholar

