
“Selfish Spermatogonial Selection”: A Novel Mechanism for the Association Between Advanced Paternal Age and Neurodevelopmental Disorders
Abstract
There is robust evidence from epidemiological studies that the offspring of older fathers have an increased risk of neurodevelopmental disorders, such as schizophrenia and autism. The authors present a novel mechanism that may contribute to this association. Because the male germ cell undergoes many more cell divisions across the reproductive age range, copy errors taking place in the paternal germline are associated with de novo mutations in the offspring of older men. Recently it has been recognized that somatic mutations in male germ cells that modify proliferation through dysregulation of the RAS protein pathway can lead to within-testis expansion of mutant clonal lines. First identified in association with rare disorders related to paternal age (e.g., Apert syndrome, achondroplasia), this process is known as “selfish spermatogonial selection.” This mechanism favors propagation of germ cells carrying pathogenic mutations, increasingly skews the mutational profile of sperm as men age, and enriches de novo mutations in the offspring of older fathers that preferentially affect specific cellular signaling pathways. This mechanism not only offers a parsimonious explanation for the association between advanced paternal age and various neurodevelopmental disorders but also provides insights into the genetic architecture (role of de novo mutations), neurobiological correlates (altered cell cycle), and some epidemiological features of these disorders. The authors outline hypotheses to test this model. Given the secular changes for delayed parenthood in most societies, this hypothesis has important public health implications.
Occasionally, seemingly distant fields of research intersect and can catalyze new discoveries. In this review we outline how divergent clues from epidemiological and genetic studies in autism and schizophrenia research can be integrated with an innovative hypothesis originally proposed to explain the relatively high apparent mutational rate associated with congenital disorders such as Apert syndrome, achondroplasia, and “RASopathies,” syndromes associated with the RAS family of proteins, which are involved in intracellular signal transmission. The “selfish spermatogonial selection” hypothesis predicts that somatic mutations that promote clonal expansion within the male germline progenitors will skew the influence of paternally derived de novo mutations. The proposed mechanism may provide a parsimonious explanation for diverse findings related to neurodevelopmental disorders such as schizophrenia and autism. A set of testable predictions are proposed to evaluate this hypothesis.
Key Differences Between Male and Female Gamete Production
Advanced Paternal Age and Mental Disorders
It was noted over 30 years ago that schizophrenia occurs more frequently in the offspring of older fathers (8). However, this observation had been largely forgotten until Malaspina and colleagues (9) suggested that this finding may be related to copy error mutations in the male germline. In a large Israeli birth cohort, they found that paternal age was a significant predictor of schizophrenia risk.
There is a growing body of epidemiological research linking advanced paternal age with other neuropsychiatric disorders and brain-related outcomes. These include autism and related spectrum disorders (11–13), bipolar disorder (14), epilepsy (15), sporadic Alzheimer’s disease (16), obsessive-compulsive disorder (17), and impaired childhood cognitive ability (18). Thus, while the evidence base is incomplete, the data suggest that advanced paternal age is associated with a wide range of brain-related adverse health outcomes (i.e., the exposure is nonspecific with respect to health outcomes). Within a community-based sample of healthy children, paternal age was found to be significantly associated with cortical gray (but not white) matter volume (19). Finally, evidence from mouse models shows that the offspring of older sires have altered behavioral outcomes (20, 21), altered brain structure (20), and increased de novo CNVs (22).
More recently, studies have suggested that grandpaternal age (specifically in the mother’s line) may also contribute to the risk of neurodevelopmental disorders (23, 24). This may be understood when one considers that phenotypes associated with predisposing mutations tend to be more pronounced in males than in females, therefore effectively leading to the skipping of generations. This grandpaternal age effect suggests that mutations can accumulate across generations. Although mutations with larger phenotypic impacts are less likely to be transmitted to subsequent generations (for instance, individuals with schizophrenia and autism have markedly reduced fertility and fecundity), mutations associated with more subtle phenotypes or variable penetrance may be transmitted to offspring and thus contribute to the “mutational burden” in subsequent generations (discussed in the following). In light of the secular trend of delayed parenthood (25), Crow suggested that paternal-age-related mutations could contribute to a transgenerational “mutational time bomb” (1).
Paternal Age Effect Mutations and Selfish Selection in the Testis
Although the differences between male and female gamete production provide a plausible explanation for the link between advanced paternal age and disease risk, recent evidence suggests that there could be more to this association than the simple accumulation of random copy errors during spermatogenesis. We have previously defined paternal age effect mutations as a small group of well-characterized genetic alterations that are associated with rare Mendelian dominant disorders and have unusual characteristics (5, 26). The best-known examples involve specific activating point mutations in the fibroblast growth factor receptor genes FGFR2 (associated with Apert and other craniosynostosis syndromes) and FGFR3 (associated with achondroplasia and other short-limbed bone dysplasias) and in several members of the RTK/RAS and associated MAPK signaling pathways (involving RAS proteins and mitogen-activated protein kinase), such as PTPN11/SHP2 and HRAS (associated with neurocardiofaciocutaneous syndromes, such as Noonan and Costello syndromes, which are collectively termed “RASopathies”) (5). In the majority of cases, these point mutations 1) are acquired de novo, 2) exhibit a near-exclusive paternal origin, 3) have a high apparent germline mutation rate (up to 1,000-fold above background for Apert syndrome and achondroplasia, according to birth prevalence), and 4) are associated epidemiologically with a significant paternal age effect (with unaffected fathers approximately 2 to 7 years older on average than the population mean). Because of these features, paternal age effect mutations provide an excellent model for studying the effect of advanced paternal age at a molecular level.
Levels of specific pathogenic mutations associated with paternal age effect disorders in the sperm of healthy men of different ages (26–28) and in dissected whole testes (29–31) have been directly quantified. The results gathered from these technically challenging experiments showed that paternal age effect mutations are detected in the sperm (but not in the blood) of most men, that the measured levels account for the birth prevalence of the associated disorders, and that the levels positively correlate with the donor’s age, in line with the observed paternal age effect. To explain these findings, we previously suggested (5, 26, 27) that
At a cellular level, selfish selection appears to operate through a mechanism that shares many of its features with oncogenesis. Indeed, all genes associated with the paternal age effect are known oncogenes that are able to promote tumorigenesis in different cellular contexts (5). Strikingly, at a molecular level, there is a common denominator to selfish selection. Paternal age effect genes cluster within a single pathway, the receptor tyrosine kinase/RAS (RTK/RAS) signaling cascade (Figure 1). Not surprisingly, this pathway is a key determinant of spermatogonial cell self-renewal, and its role in controlling the balance of proliferation/differentiation during mammalian spermatogenesis has been well documented in murine models (5). This effect is mediated through control of several different transduction pathways downstream of RAS, shown in Figure 1. The best-characterized pathways include the MAPK pathway (shown in red in Figure 1), for which ERK1/2 is a downstream effector, and the PI3K/AKT cascade (shown in green), for which mTOR is a downstream effector (see Figure 1 for definitions of abbreviations). The RAS and RAS-related RHEB (RAS homolog enriched in brain) signaling pathways, which are commonly dysregulated in cancer, are widely connected with many essential cellular processes and have a key role in neuronal functioning, including synaptic plasticity (33, 34).
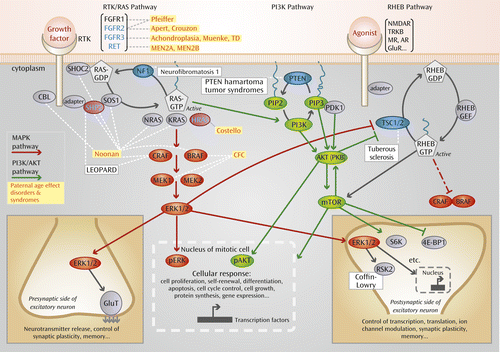
a Abbreviations: AKT, a serine/threonine-specific protein kinase, also known as protein kinase B (PKB); AR, adrenergic receptor; BRAF, a member of the RAF (rapidly accelerated fibrosarcoma) kinase family; CBL, protein encoded by Cbl gene (named after Casitas B-lineage lymphoma); CFC, cardiofaciocutaneous syndrome; CRAF, a member of the RAF (rapidly accelerated fibrosarcoma) kinase family; 4E-BP1, 4E binding protein 1; ERK, extracellular-signal-regulated kinase; FGFR, fibroblast growth factor receptor gene; GDP, guanosine 5′-diphosphate; GEF, guanine nucleotide exchange factor; GluR, glutamate receptor; GluT, glutamate transporter; GTP, guanosine 5′-triphosphate; HRAS, enzyme encoded by the Harvey RAS-1 gene; KRAS, protein encoded by the Kirsten RAS oncogene; MAPK, mitogen-activated protein kinase; MEK, a protein kinase (“mitogen-activated protein kinase/extracellular-signal-regulated kinase”); MEN, multiple endocrine neoplasia; MR, mineralocorticoid receptor; mTOR, mammalian target of rapamycin (a serine/threonine protein kinase); NMDAR, N-methyl-d-aspartate receptor; NRAS, enzyme encoded by the NRAS gene, which is associated with neuroblastoma; pAKT, phosphorylated form of AKT; PDK, phosphoinositide-dependent kinase; pERK, phosphorylated form of ERK; PI3K, phosphatidylinositide 3-kinase; PIP, prolactin-inducible protein; PKB, protein kinase B; PTEN, protein encoded by the phosphatase and tensin homolog gene; RAS, a family of proteins involved in intracellular signal transmission (from “rat sarcoma”); RET, “rearranged during transfection” gene; RHEB, RAS homolog enriched in brain; RSK2, ribosomal S6 kinase; RTK, receptor tyrosine kinase; S6K, S6 kinase; SHOC, protein encoded by “suppressor of clear homolog in C. elegans” gene; SHP2, cytosolic SH2 domain containing protein tyrosine phosphatase; SOS, protein encoded by “son of sevenless” gene; TD, thanatophoric dysplasia; TRKB, tyrosine-related kinase B; TSC, tuberous sclerosis complex. LEOPARD is a mnemonic of characteristic features.
b See references 5 and 32 for more details on these pathways. Germline disorders associated with mutations in specific genes along these pathways are indicated in boxes. The gene products that belong to the class affected by paternal age (as defined in text) are in blue, and yellow boxes indicate related disorders. Congenital disorders associated with the RAS family are known as “RASopathies.” They include the Noonan, Costello, LEOPARD, and CFC syndromes and are caused by mutations in the RTK/RAS and MAPK pathways. Other proteins in the pathway and associated germline disorders for which evidence of direct involvement in the process of selfish spermatogonial selection and paternal age effect is still lacking are indicated in black. Known tumor-suppressor genes in cancer are indicated by blue ovals. The RAS pathway is involved in many cellular processes, and some of the consequences of pathway activation are illustrated in the case of transduction occurring in a mitotically active cell (bottom, middle) or during neurotransmission and/or synaptic plasticity (bottom, left and right). Translocation of phosphorylated forms of ERK (pERK) or AKT (pAKT) into the nucleus of a mitotic cell triggers many different cellular responses, such as cell growth, proliferation, differentiation, motility, and apoptosis. Within excitatory neurons, a few examples of cellular responses triggered by the RAS or RHEB pathway are illustrated and involve molecules such as ribosomal S6 kinase (S6K and RSK2) and eukaryotic translation initiation factor 4E-BP1.
Although this paradigm is based on a handful of paternal age effect mutations of unusually large effect, it is likely that other selfish pathways operate in the context of the aging testis. For example, the signaling networks triggered by oncogenic activation of RAS are highly complex and involve crosstalk between multiple pathways (32). In particular, we might expect other well-characterized oncogenic pathways, such as PI3K/AKT and their effectors, which are key regulators of spermatogonial cell proliferation, to participate in this process. Thus, while this article will focus on the canonical RAS-related pathways, we speculate that mutations in any gene that is expressed in spermatogonial cells and whose function involves regulation of homeostasis through control of proliferation/differentiation balance or cell cycle are potential targets of selfish selection (5). Similarly, selfish mutations are unlikely to be limited to point mutations with strong gain-of-function properties, such as those described so far for paternal age effect mutations; we predict that other genetic alterations, such as CNVs, small indels, or regulatory mutations, will also be targets of selfish selection.
Genetic Architecture of Neurocognitive Disorders and Selfish Selection
While not all de novo mutations associated with neuropsychiatric disorders are related to paternal age (for instance, 22q11.2 deletions are not), we note with interest that there is overlap between the molecular pathways that have been identified during exome/CNV screens for schizophrenia and autism and those implicated in selfish selection in the testis. In their large study of CNVs in autism, Pinto et al. described a noticeable enrichment of rare pathogenic CNVs—both inherited and de novo—disrupting genes involved in cellular proliferation, projection and motility, and RAS signaling (35). Studies analyzing proband-parent trios in autism identified de novo mutations in genes clustering within molecular pathways implicated in tumorigenesis and control of early development, such as RAS, p53, and β-catenin pathways (35–39). There is also evidence suggesting that individuals diagnosed with RASopathies have more difficulties in adaptive functioning (40) and show autistic traits (41). Thus, within the fields of autism and schizophrenia research, there has been an unexpected overlap between 1) candidate pathogenic variants in genes that are being discovered through genetic studies and 2) genes involved in tumorigenesis as well as selfish selection in the testis.
With respect to genetic risk factors, researchers have previously noted that some candidate genes linked to schizophrenia are also mutated in various cancers. These include the gene for protein kinase B (PKB, also known as AKT), which is commonly mutated in cancer, and the gene for neuregulin-1, which is associated with breast cancer, B cell leukemia/lymphoma, and multiple myeloma (42, 43). Moreover, genome-wide association studies (GWAS) in schizophrenia have identified strong signals near the paternal age effect genes FGFR1 (44) and FGFR2 (45), and there is convergent evidence linking disruption of AKT (42) and fibroblast growth factor signaling with schizophrenia (46) as well as linking cancer-related pathways (e.g., PI3K/AKT, PTEN) and autism (47).
Weak Selfish Mutations and Common Diseases
Although sperm studies of paternal age effect mutations have highlighted that selfish selection relies on the activation of the RAS pathway, the role of RAS is clearly not limited to processes occurring in the testis. This pathway is a crucial mediator of intracellular signaling in many contexts and is recruited at different times in development to execute distinct functions, including cell survival, proliferation, migration, differentiation, and motility. Hence, dysregulation of the RAS pathway in the adult testis is predicted to have consequences that extend far beyond rare dominant disorders. To consider whether selfish selection could have a more pervasive role in complex disorders, we need to compare the effect of the strength of selfish selection occurring in the testis (which promotes clonal expansion and leads to relative enrichment in mutant sperm) and the impact that given mutations are anticipated to have on the fitness of the offspring who inherit the mutation upon germline transmission. Because of the strong pathogenic germline phenotypes they cause, paternal age effect mutations—such as those commonly described in cancer and those associated with paternal age effect disorders—will be associated with lethal or deleterious phenotypes that are unlikely to segregate in the population or have a long-term impact on disease burden (Figure 2 top; red mutations). However, selfish mutations providing a weaker selective advantage in the testes (which will be enriched to a lesser extent in sperm) are predicted to be associated with more subtle phenotypes (such as variants with low/variable penetrance or susceptibility). The difference in impact is important: these mild selfish mutations are a potential source of heritable variations that could contribute to the genetic burden of common diseases (Figure 2, orange mutations) and to genetic heterogeneity (Figure 2, yellow mutations).
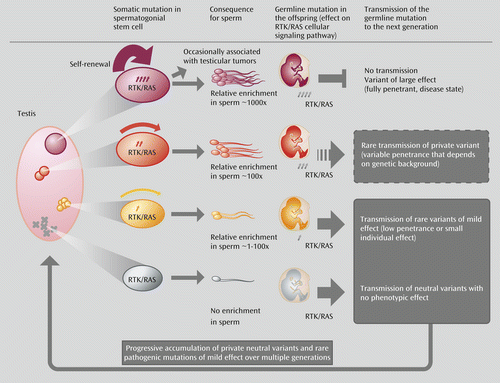
a Abbreviations: RAS, a family of proteins involved in intracellular signal transmission (from “rat sarcoma”); RTK, receptor tyrosine kinase. A “private” variant is a mutational hit that is likely to be serendipitous and therefore is anticipated to be essentially unique.
b The pink oval (left) represents the testis of an aging man in which mutations (represented by X or circles) have occurred randomly during the recurrent rounds of replication required for spermatogenesis. In the gray section (bottom of diagram), functionally neutral mutations are not enriched in spermatogonial progenitors and are associated with a very low risk of transmission of individual genetic lesions in the offspring. In the red section (top of diagram), the mutant spermatogonial progenitor carries a “strong effect” mutation that is associated with overt dysregulation of the RTK/RAS pathway, such as a typical oncogenic mutation. These paternal age effect mutations confer a strong selective advantage (represented by the thickness of the red arrow) to the mutant spermatogonial stem cell, leading over time to the formation of large clones and relative enrichment in mutant sperm. Upon germline transmission to the offspring, strong paternal age effect mutations can cause neonatal lethality or disease phenotypes and are eliminated by purifying selection, as affected individuals are unlikely to reproduce. The orange and yellow sections (middle of the diagram) depict intermediate scenarios for mutations with milder selective advantage (i.e., mutation of variable penetrance, weak gain of function) that are enriched over time in spermatogonial stem cells to a lesser extent (>1- to 100-fold). Unlike the mutations with strong effects (red), mildly pathogenic mutations are potentially transmissible over many generations, contributing to genetic heterogeneity and variable expressivity of disease phenotypes.
Does the Selfish Selection Hypothesis Have Explanatory Power?
Clues Linking Paternal Age, Cancer, and Neurodevelopmental Disorders
There is some evidence (5) linking advanced paternal age with an increased risk of a range of cancer types (e.g., childhood brain tumors and leukemia, non-Hodgkin lymphoma, breast and testicular cancer). If advanced paternal age is associated with risk of both schizophrenia and cancer, then it might be predicted that cancer would be more common in schizophrenia patients. However, the epidemiological data on this issue are inconsistent (48–50), and the effects seem to vary depending on the cancer types considered (51). On balance, the studies show that the incidence of cancer in individuals with schizophrenia is lower than expected (52). Moreover, the lower risk for several types of cancer has also been observed in unaffected family members of schizophrenia patients, providing further support for genetic protection against cancer in families with schizophrenia (48, 52, 53). Given the overlap between the molecular pathways involved in brain development and those associated with tumorigenesis, mutations that have been enriched in germ cells through mechanisms such as selfish selection are expected to modify the risk of both neurocognitive diseases and cancer when inherited by the offspring. As we learn more about the genetic basis of cancer and neuropsychiatric disorders, the precise relationship between these two broad categories of disorders may be clarified.
Clues Related to Minor Physical Anomalies, the Cell Cycle, and Synaptic Plasticity
The selfish selection hypothesis emerged from a group of congenital disorders that are often characterized by prominent craniofacial abnormalities (5). It has long been established that minor physical anomalies are more prevalent in individuals with schizophrenia (54) or autism (55).
Properties of cell lines such as those obtained from “neurosphere-derived” cells generated from olfactory mucosal biopsies from patients with schizophrenia and from healthy comparison subjects have been examined (56). The patient-derived cells showed significant dysregulation of genes involved in cell cycle control (in particular, various cyclins and several cyclin-dependent kinase 4 (CDK4) inhibitors controlling the G1/S phase transition in the cell cycle). Changes in expression of genes and/or proteins involved in the cell cycle are particularly interesting because increased cell proliferation has previously been demonstrated in olfactory biopsies from schizophrenia patients (57, 58). Induced pluripotent stem cells from individuals with LEOPARD syndrome (one of the RASopathies) have provided clues about the impact of these mutations on different derived cell types (59). It will be of interest to establish whether fibroblast-derived induced pluripotent stem (iPS) cells from individuals with neuropsychiatric disorders can be used to identify signaling pathways contributing to disease phenotype (60).
There is a growing body of evidence indicating that genes that were once thought to be involved only in cell cycle control also ‘moonlight’ in a range of critical functions in postmitotic neurons (see review by Frank and Tsai [61]). For example, cyclins have been implicated in axonal growth (62) and dendritic morphogenesis (63). Synaptic function is also being linked to genes better known for their role in cancer, cell cycle control, and differentiation (64, 65) (Figure 1). These mechanisms may represent important clues for understanding the links between schizophrenia and cancer (66).
An Integrative Model and Testable Hypotheses
As mechanisms such as selfish selection operate in the aging human testis, it is anticipated that over the reproductive life of the male, mutations in germ cell progenitors that influence cell cycle/growth control pathways (in particular, within the RTK/RAS signaling cascade) will promote spermatogonial proliferation and result in relative enrichment of mutant sperm over time. Given that whole-genome sequencing data have confirmed that each newborn carries 30–100 novel point mutations in its genome (7), there is a possibility that a proportion of the paternally derived mutations will preferentially carry alterations in genes that fall within these selfish pathways. These mutations would have variable consequences for the phenotype and transmission to the next generation (Figure 2).
Conveniently, this hypothesis may provide insights into some of the more puzzling features of neurodevelopmental disorders, such as the nature of the “missing heritability” associated with these genetic diseases (for more details, see reference 67). Selfish selection is predicted to promote a weak bias favoring fertilization by sperm carrying mild pathogenic selfish alleles. Although this process occurs at each generation, the exact nature of the mutational hit involved is likely to be serendipitous and therefore is anticipated to be essentially unique (“private”) and would be identifiable only during a genome-wide unbiased sequencing screen (i.e., not during GWAS, which assess only common variants). Thus, this hypothesis aids formulation of a parsimonious framework to understand 1) the association between advanced paternal age and various neurocognitive disorders and some cancers, 2) the complex genetic architecture of neurodevelopmental disorders and the important role of de novo/private mutations clustering in key signaling pathways, 3) the high heritability, comorbidity, and high prevalence of these deleterious conditions associated with low fertility, and 4) the various clinical and epidemiological clues related to dysmorphogenesis, cell cycle properties, and altered risk of comorbid cancer.
We readily acknowledge many limitations of this broad model. Not all neurodevelopmental disorders are caused by de novo mutations, nor do we propose that all genetic factors related to these disorders are restricted to RTK/RAS and related pathways. However, we hope that the selfish selection hypothesis will be useful as a catalyst for debate, data interpretation, and new hypothesis-driven research. Following is a list of testable hypotheses based on this mechanism.
| 1. | Whole-genome sequencing of family trios has demonstrated a significant excess of paternally derived de novo mutations (7). Although, as a whole, the number of de novo mutations correlates with the father’s age, selfish selection predicts that mutations dysregulating signaling pathways such as RTK/RAS and their downstream effectors will be overrepresented in the offspring of older fathers. The relationship between paternal age and de novo mutational load should therefore be examined independently for different categories of genetic alterations. Thus, the correlation between paternal age and de novo mutations in genes falling in selfish pathways will be greater than for neutral mutations. | ||||
| 2. | Sequencing of multiple individual sperm from men of different ages will confirm the correlation between paternal age and excess of de novo mutations dysregulating selfish pathways such as RTK/RAS. | ||||
| 3. | Susceptibility variants for schizophrenia and autism risk will be significantly more likely to affect pathways involved in the cell cycle and the RTK/RAS signaling cascade than would be predicted by chance. | ||||
| 4. | Patient-derived cell lines (e.g., olfactory neurospheres, iPS cell lines) from patients with disorders related to paternal age, such as schizophrenia and autism, will be more likely to display phenotypic changes related to the RTK/RAS signaling pathway than would be predicted by chance. | ||||
Conclusions
So far, data have indicated that the RTK/RAS pathway is a crucial mediator of selfish selection. As we learn more about the mechanisms controlling spermatogonial proliferation, the preceding hypotheses should expand beyond the canonical RTK/RAS cascade and include other signaling pathways that are potential targets for selfish selection. Combining this knowledge with our understanding of the genetic networks involved in neurodevelopmental disorders should help define the extent of the overlap between the two processes. Ultimately, this will provide insights into a broader research question: Are mutations associated with these complex disorders part of a “normal” process that produces genetic variation with each generation and are therefore the unavoidable consequence of a selfish mechanism originating in the aging testis?
The selfish spermatogonial selection hypothesis provides an example of the creative intersection of diverse research fields—in this case, epidemiology, animal models, molecular and clinical genetics, genomics, oncology, cell biology, and psychiatry. From a public health perspective, the impact of advanced paternal age on health outcomes may become more apparent over time, as parenthood is delayed in many societies as a result of educational and cultural factors (reviewed in reference 25). Because these mutations are proposed to accumulate over several generations, mechanisms related to selfish selection could have far-reaching consequences for the health of future generations.
1 : The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 2000; 1:40–47Crossref, Medline, Google Scholar
2 : A new study challenges the current belief of a high human male:female mutation ratio. Trends Genet 2000; 16:525–526Crossref, Medline, Google Scholar
3 : De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J Med Genet 2011; 48:776–778Crossref, Medline, Google Scholar
4 : A direct characterization of human mutation based on microsatellites. Nat Genet 2012; 44:1161–1165Crossref, Medline, Google Scholar
5 : Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 2012; 90:175–200Crossref, Medline, Google Scholar
6 : Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 2012; 150:402–412Crossref, Medline, Google Scholar
7 : Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012; 488:471–475Crossref, Medline, Google Scholar
8 : Raised parental age in psychiatric patients: evidence for the constitutional hypothesis. Br J Psychiatry 1979; 134:169–177Crossref, Medline, Google Scholar
9 : Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry 2001; 58:361–367Crossref, Medline, Google Scholar
10 : Meta-analysis of paternal age and schizophrenia risk in male versus female offspring. Schizophr Bull 2011; 37:1039–1047Crossref, Medline, Google Scholar
11 : Advancing paternal age and autism. Arch Gen Psychiatry 2006; 63:1026–1032Crossref, Medline, Google Scholar
12 : Independent and dependent contributions of advanced maternal and paternal ages to autism risk. Autism Res 2010; 3:30–39Crossref, Medline, Google Scholar
13 : Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies. Mol Psychiatry 2011; 16:1203–1212Crossref, Medline, Google Scholar
14 : Advancing paternal age and bipolar disorder. Arch Gen Psychiatry 2008; 65:1034–1040Crossref, Medline, Google Scholar
15 : Paternal age and epilepsy in the offspring. Eur J Epidemiol 2005; 20:1003–1005Crossref, Medline, Google Scholar
16 : Paternal age is a risk factor for Alzheimer disease in the absence of a major gene. Neurogenetics 1998; 1:277–280Crossref, Medline, Google Scholar
17 : Advanced paternal age increases the risk of schizophrenia and obsessive-compulsive disorder in a Chinese Han population. Psychiatry Res 2012; 198:353–359Crossref, Medline, Google Scholar
18 : Advanced paternal age is associated with impaired neurocognitive outcomes during infancy and childhood. PLoS Med 2009; 6:e40Crossref, Medline, Google Scholar
19 : Parental age effects on cortical morphology in offspring. Cereb Cortex 2012; 22:1256–1262Crossref, Medline, Google Scholar
20 : Advanced paternal age is associated with alterations in discrete behavioural domains and cortical neuroanatomy of C57BL/6J mice. Eur J Neurosci 2010; 31:556–564Crossref, Medline, Google Scholar
21 : Advancing paternal age is associated with deficits in social and exploratory behaviors in the offspring: a mouse model. PLoS ONE 2009; 4:e8456Crossref, Medline, Google Scholar
22 : Increased de novo copy number variants in the offspring of older males. Transl Psychiatry 2011; 1:e34Crossref, Medline, Google Scholar
23 : Advanced paternal and grandpaternal age and schizophrenia: a three-generation perspective. Schizophr Res 2011; 133:120–124Crossref, Medline, Google Scholar
24 : Autism risk develops across generations: a population based study of advancing grandpaternal and paternal age. JAMA Psychiatry (in press)Google Scholar
25 : Advanced paternal age: how old is too old? J Epidemiol Community Health 2006; 60:851–853Crossref, Medline, Google Scholar
26 : Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet 2009; 41:1247–1252Crossref, Medline, Google Scholar
27 : Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 2003; 301:643–646Crossref, Medline, Google Scholar
28 : The ups and downs of mutation frequencies during aging can account for the Apert syndrome paternal age effect. PLoS Genet 2009; 5:e1000558Crossref, Medline, Google Scholar
29 : The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol 2007; 5:e224Crossref, Medline, Google Scholar
30 : A germ-line-selective advantage rather than an increased mutation rate can explain some unexpectedly common human disease mutations. Proc Natl Acad Sci USA 2008; 105:10143–10148Crossref, Medline, Google Scholar
31 : Positive selection for new disease mutations in the human germline: evidence from the heritable cancer syndrome multiple endocrine neoplasia type 2B. PLoS Genet 2012; 8:e1002420Crossref, Medline, Google Scholar
32 : Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295–308Crossref, Medline, Google Scholar
33 : MAP’ing CNS development and cognition: an ERKsome process. Neuron 2009; 61:160–167Crossref, Medline, Google Scholar
34 : Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 2011; 49:296–305Crossref, Medline, Google Scholar
35 : Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466:368–372Crossref, Medline, Google Scholar
36 ;
37 : Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012; 485:246–250Crossref, Medline, Google Scholar
38 : De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012; 17:142–153Crossref, Medline, Google Scholar
39 : Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011; 70:886–897Crossref, Medline, Google Scholar
40 : Effects of germline mutations in the Ras/MAPK signaling pathway on adaptive behavior: cardiofaciocutaneous syndrome and Noonan syndrome. Am J Med Genet A 2010; 152A:591–600Crossref, Medline, Google Scholar
41 : Autism traits in the RASopathies (abstract), in Abstracts of the 62nd Annual Meeting of the American Society of Human Genetics. Bethesda, Md, ASHG, 2012, program number 99 (http://abstracts.ashg.org/cgi-bin/2012/ashg12s?pgmnr=99&sort=ptimes&sbutton=Detail&absno=120120866&sid=238158)Google Scholar
42 : AKT/GSK3 signaling pathway and schizophrenia. Front Mol Neurosci 2012; 5:33Crossref, Medline, Google Scholar
43 : The neuregulin-I/ErbB signaling system in development and disease. Adv Anat Embryol Cell Biol 2007; 190:1–65Crossref, Medline, Google Scholar
44 : Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat Genet 2011; 43:1224–1227Crossref, Medline, Google Scholar
45 ;
46 : Fibroblast growth factors in schizophrenia. Schizophr Bull 2010; 36:1157–1166Crossref, Medline, Google Scholar
47 : Autism and cancer risk. Autism Res 2011; 4:302–310Crossref, Medline, Google Scholar
48 : Incidence of cancer among persons with schizophrenia and their relatives. Arch Gen Psychiatry 2001; 58:573–578Crossref, Medline, Google Scholar
49 : Schizophrenia and breast cancer incidence: a systematic review of clinical studies. Schizophr Res 2009; 114:6–16Crossref, Medline, Google Scholar
50 : Cancer mortality in patients with psychiatric diagnoses: a higher hazard of cancer death does not lead to a higher cumulative risk of dying from cancer. Soc Psychiatry Psychiatr Epidemiol (Epub ahead of print, Oct 27, 2012)Google Scholar
51 : No paradox, no progress: inverse cancer comorbidity in people with other complex diseases. Lancet Oncol 2011; 12:604–608Crossref, Medline, Google Scholar
52 : Cancer incidence in patients with schizophrenia and their first-degree relatives—a meta-analysis. Acta Psychiatr Scand 2008; 117:323–336Crossref, Medline, Google Scholar
53 : Incidence of cancer in patients with schizophrenia and their first-degree relatives: a population-based study in Sweden. Schizophr Bull (Epub ahead of print, April 20, 2012)Google Scholar
54 : Physical manifestations of neurodevelopmental disruption: are minor physical anomalies part of the syndrome of schizophrenia? Schizophr Bull 2009; 35:425–436Crossref, Medline, Google Scholar
55 : Minor physical anomalies in children with autism spectrum disorders. Autism 2011; 15:746–760Crossref, Medline, Google Scholar
56 : Disease-specific, neurosphere-derived cells as models for brain disorders. Dis Model Mech 2010; 3:785–798Crossref, Medline, Google Scholar
57 : Altered adhesion, proliferation and death in neural cultures from adults with schizophrenia. Schizophr Res 1999; 40:211–218Crossref, Medline, Google Scholar
58 : Cell cycle alterations in biopsied olfactory neuroepithelium in schizophrenia and bipolar I disorder using cell culture and gene expression analyses. Schizophr Res 2006; 82:163–173Crossref, Medline, Google Scholar
59 : Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010; 465:808–812Crossref, Medline, Google Scholar
60 : Modeling psychiatric disorders through reprogramming. Dis Model Mech 2012; 5:26–32Crossref, Medline, Google Scholar
61 : Alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron 2009; 62:312–326Crossref, Medline, Google Scholar
62 : Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 2006; 442:471–474Crossref, Medline, Google Scholar
63 : A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 2009; 136:322–336Crossref, Medline, Google Scholar
64 : Common pathways for growth and for plasticity. Curr Opin Neurobiol 2012; 22:405–411Crossref, Medline, Google Scholar
65 : N-methyl-D-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J 2002; 21:2139–2148Crossref, Medline, Google Scholar
66 : Do shared mechanisms underlying cell cycle regulation and synaptic plasticity underlie the reduced incidence of cancer in schizophrenia? Schizophr Res 2011; 130:282–284Crossref, Medline, Google Scholar
67 : Missing heritability: paternal age effect mutations and selfish spermatogonia (letter). Nat Rev Genet 2010; 11:589Crossref, Medline, Google Scholar

