
Deficits in Transcriptional Regulators of Cortical Parvalbumin Neurons in Schizophrenia
Abstract
Objective
In schizophrenia, alterations within the prefrontal cortical GABA system appear to be most prominent in neurons that contain parvalbumin or somatostatin but not calretinin. The transcription factors Lhx6 and Sox6 play critical roles in the specification, migration, and maturation of parvalbumin and somatostatin neurons, but not calretinin neurons, and continue to be strongly expressed in this cell type-specific manner in the prefrontal cortex of adult humans. The authors investigated whether Lhx6 and/or Sox6 mRNA levels are deficient in schizophrenia, which may contribute to cell type-specific disturbances in cortical parvalbumin and somatostatin neurons.
Method
The authors used quantitative PCR and in situ hybridization with film and grain counting analyses to quantify mRNA levels in postmortem samples of prefrontal cortex area 9 of 42 schizophrenia subjects and 42 comparison subjects who had no psychiatric diagnoses in life, as well as antipsychotic-exposed monkeys.
Results
In schizophrenia subjects, the authors observed lower mRNA levels for Lhx6, parvalbumin, somatostatin, and glutamate decarboxylase (GAD67; the principal enzyme in GABA synthesis), but not Sox6 or calretinin. Cluster analysis revealed that a subset of schizophrenia subjects consistently showed the most severe deficits in the affected transcripts. Grain counting analyses revealed that some neurons that normally express Lhx6 were not detectable in schizophrenia subjects. Finally, lower Lhx6 mRNA levels were not attributable to psychotropic medications or illness chronicity.
Conclusions
These data suggest that in a subset of individuals with schizophrenia, Lhx6 deficits may contribute to a failure of some cortical parvalbumin and somatostatin neurons to successfully migrate or develop a detectable GABA-ergic phenotype.
Prefrontal cortex-related cognitive impairments in schizophrenia have been linked to disturbances in the inhibitory system, such as deficits in the GABA synthesizing enzyme glutamate decarboxylase (GAD67) (1–6). Alterations in cortical GABA neurons are most prominent in the subsets that contain the calcium-binding protein parvalbumin or the neuropeptide somatostatin but not in the subset that expresses the calcium-binding protein calretinin. For example, mean levels of parvalbumin, somatostatin, and GAD67 mRNAs in the prefrontal cortex have been consistently reported to be lower in cohorts of schizophrenia subjects, but not all schizophrenia subjects in each cohort had lower levels than their matched comparison subjects (5, 7–10). The pathogenetic mechanisms that lead to the molecular pathology of specific GABA cell types that are prominent in a subset of schizophrenia subjects are not known; however, factors related to the developmental origin of the different GABA neuron subpopulations may play a role.
Indeed, the specification of cortical GABA neurons into distinct subpopulations is related to the location of their origination and is regulated by cell type-specific transcription factors. For example, parvalbumin and somatostatin neurons originate from the medial ganglionic eminence of the subpallium in humans and rodents (11–16), whereas calretinin neurons derive from the subventricular zone of the dorsal pallium, at least in primates and humans (14–18). Furthermore, during the prenatal period, certain transcription factors (e.g., Lhx6 and Sox6) selectively regulate the ontogeny (i.e., cell type specification, tangential migration, and maturation) of parvalbumin and somatostatin neurons, but not calretinin neurons (19–22). A complete loss of Lhx6 or Sox6 in prenatal periods leads to slowed tangential migration to the cerebral cortex, impeded differentiation into parvalbumin and somatostatin neurons, and altered GAD67 levels (19–22). These lines of evidence suggest that in schizophrenia, early developmental disturbances, such as altered expression of cell type-specific transcription factors, may lead to persisting deficits that predominantly affect parvalbumin and somatostatin neurons, but not calretinin neurons.
Although it is not feasible to directly study embryonic cortical GABA neuron ontogeny in schizophrenia, some postmortem studies of adult schizophrenia subjects have provided evidence suggestive of arrested migration of somatostatin neurons (23) and of failure of parvalbumin neurons to develop a GABA-ergic phenotype (7). Interestingly, Lhx6 and Sox6 continue to be selectively and robustly expressed by virtually all parvalbumin and somatostatin neurons in adult cortex of humans and rodents (13, 19, 21, 22, 24). In this study, we sought to determine whether mRNA levels for Lhx6 and Sox6 in postmortem human prefrontal cortex are deficient in schizophrenia and whether such deficits are especially prominent in schizophrenia subjects with clear deficits in parvalbumin, somatostatin, and GAD67 mRNAs.
Method
Human Subjects
Brain specimens were obtained during routine autopsies conducted at the Allegheny County Medical Examiner’s Office after consent was obtained from next of kin. An independent committee of experienced research clinicians made consensus DSM-IV (25) diagnoses for each subject using structured interviews with family members and review of medical records (26), and the absence of psychiatric diagnoses was confirmed in comparison subjects. To control for experimental variance, subjects with schizophrenia or schizoaffective disorder (N=42) were matched individually to one comparison subject for sex and as closely as possible for age (see Table S1 in the data supplement that accompanies the online edition of this article), as previously described (6, 26, 27), and samples from subjects in a pair were processed together throughout all stages of the study. Some subject pairs had previously been studied for parvalbumin (N=19), somatostatin (N=23), and calretinin (N=19) mRNA levels, mostly by in situ hybridization (see Table S1) (7–9, 28). The mean age, postmortem interval, freezer storage time, brain pH, and RNA integrity number (RIN; Agilent Bioanalyzer, Santa Clara, Calif.) did not differ between subject groups (Table 1), and each subject had a RIN ≥7.0. All procedures were approved by the University of Pittsburgh Committee for the Oversight of Research Involving the Dead as well as the university’s Institutional Review Board.
| Measure | Comparison Group (N=42) | Schizophrenia Group (N=42) | ||
|---|---|---|---|---|
| N | % | N | % | |
| Male | 31 | 74 | 31 | 74 |
| White | 34 | 81 | 29 | 69 |
| Black | 8 | 19 | 13 | 31 |
| Mean | SD | Mean | SD | |
| Age (years) | 48 | 13 | 47 | 13 |
| Postmortem interval (hours) | 17.8 | 5.9 | 18.1 | 8.7 |
| Freezer storage time (months) | 121 | 44 | 121 | 46 |
| Brain pH | 6.8 | 0.2 | 6.6 | 0.4 |
| RNA integrity number | 8.3 | 0.6 | 8.2 | 0.7 |
Preparation of Human Brain Tissue
Frozen tissue blocks containing the middle portion of the right superior frontal sulcus were confirmed to contain prefrontal cortical area 9 using Nissl-stained, cryostat tissue sections for each subject (2). Standardized amounts of cortical gray matter from tissue blocks were collected in TRIzol reagent in a manner that ensured minimal white matter contamination and excellent RNA preservation (8) (see the supplemental Method section in the online data supplement).
Quantitative PCR
Complementary DNA (cDNA) was synthesized from standardized dilutions of total RNA (10 ng/μl) for each subject. All primer pairs (see Table S2 in the online data supplement) demonstrated high amplification efficiency (>96%) across a wide range of cDNA dilutions and specific single products in dissociation curve analysis. Quantitative PCR (qPCR) was performed using the comparative cycle threshold (CT) method with Power SYBR Green dye and the StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, Calif.), as previously described (see the supplemental Method section in the online data supplement). Based on their stable relative expression level between schizophrenia and comparison subjects (8), three reference genes (beta actin, cyclophilin A, and glyceraldehyde-3-phosphate dehydrogenase) were used to normalize target mRNA levels. The difference in CT (dCT) for each target transcript was calculated by subtracting the geometric mean CT for the three reference genes from the CT of the target transcript (mean of four replicate measures), and differences between diagnostic groups were determined using the average dCT of each group. Because dCT represents the log2-transformed expression ratio of each target transcript to the reference genes, the relative level of the target transcript for each subject is reported as 2−dCT (8, 26, 27, 29).
In Situ Hybridization
A 988-base pair fragment corresponding to bases 1880–2867 of the human Lhx6 gene (NM_014368) was PCR-amplified, and nucleotide sequencing confirmed 100% homology for the amplified fragment to the reported sequence. Sense and antisense 35S-labeled riboprobes were generated by in vitro transcription, and hybridization procedures were performed using three sections from each subject, which were then exposed to Biomax MR film (Kodak, Rochester, N.Y.), coated with nuclear emulsion, developed, and counterstained with cresyl violet, as previously described (2, 7, 9) (see the supplemental Method section in the online data supplement). Optical density was measured in the gray matter, including individual cortical layers, using film autoradiographs, as previously described (30) (see the supplemental Method section). To evaluate Lhx6 mRNA expression at the cellular level, the numbers of silver grains generated by the 35S-labeled riboprobe in emulsion-dipped sections were counted over neurons in sampling frames placed in layers 3 and 6 (see the supplemental Method section and Figure S1 in the online data supplement). Within the frames, the number of grains was counted in a circle with a fixed diameter of 22 μm placed over each Nissl-stained nucleus. Neurons were considered to be specifically labeled if the grain density per neuron was more than five times that of background, as determined by counting the grains in the same circle placed over glial nuclei (2, 7, 9).
Antipsychotic-Exposed Monkeys
Young adult male long-tailed monkeys (Macaca fascicularis) received oral doses of haloperidol, olanzapine, or placebo (N=6 monkeys per group) twice daily for 17–27 months (31). Triads (one monkey from each of the three groups) were euthanized together on separate days. RNA was isolated from prefrontal cortical area 9, and qPCR was conducted for the same three reference genes and Lhx6 (see Table S2 in the online data supplement) with samples from all monkeys from a triad processed together on the same plate. All animal studies followed the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Statistical Analysis
Analyses of covariance (ANCOVA) were first conducted to determine whether mRNA levels were related to sex, age at death, postmortem interval, brain pH, RIN, and storage time. We found that relative somatostatin mRNA levels were related to age (F=28.7, df=1, 76, p<0.001), as previously reported (9), and that relative Lhx6 mRNA levels quantified by qPCR were related to tissue storage time (F=7.2, df=1, 76, p=0.009) (see Figure S2 in the online data supplement). No other postmortem factors were found to affect mRNA levels, and none of these factors differed between diagnostic groups (Table 1). Consequently, the ANCOVA model we report includes mRNA level as the dependent variable, diagnostic group as the main effect, subject pair as a blocking factor, and, only for relative Lhx6 mRNA levels, storage time as a covariate. Subject pairing may be considered an attempt to account for the parallel processing of tissue samples from a pair and to balance diagnostic groups for sex and age, and not a true statistical paired design. Therefore, a second ANCOVA model without subject pair as a blocking factor was also used, and both models produced similar results. Subsequent analyses of differences in mRNA levels between schizophrenia subjects grouped by substance abuse and psychotropic medications were conducted using the unpaired ANCOVA models with an alpha of 0.05.
To determine whether a subset of schizophrenia subjects express Lhx6, GAD67, parvalbumin, and somatostatin mRNAs in a distinct pattern, a cluster analysis was conducted using the levels of these four transcripts from each of the schizophrenia and comparison subjects (N=84). To account for significant effects of covariates (see Figure S2 in the online data supplement), we first adjusted somatostatin mRNA levels for each subject to the average age of all subjects at death (mean=47.6 years) and Lhx6 mRNA levels to the average storage time of all subjects’ samples (mean=120.8 months). To account for varying scales among the four mRNAs, standardized mRNA levels of storage time-adjusted Lhx6, GAD67, parvalbumin, and age-adjusted somatostatin values were computed over all subjects by subtracting the overall mean and then dividing by the overall standard deviation. Standardized mRNA levels were used to cluster all schizophrenia and comparison subjects using the average linkage method (PROC CLUSTER in SAS; SAS Institute, Cary, N.C.), and the clustering result is presented in a tree dendrogram. Standardized mRNA levels were compared among the groups arising from the clustering using ANCOVA models as described earlier. Pearson chi-square analysis was then used to determine whether a subset of schizophrenia subjects was more likely than the remaining schizophrenia subjects to have indicators of greater illness severity.
For the antipsychotic-exposed monkey study, an analysis of variance model with mRNA level as the dependent variable, treatment group as the main effect, and triad as a blocking factor was employed.
Results
Cortical Parvalbumin, Somatostatin, and Calretinin mRNA Levels in Schizophrenia
We first sought to replicate prior reports of lower average parvalbumin and somatostatin mRNA levels (5, 7–10, 28) in the prefrontal cortex in schizophrenia using qPCR. As expected, mean mRNA levels in schizophrenia subjects were lower for parvalbumin (−22%; F=17.5, df=1, 41, p<0.001) and somatostatin (−36%; F=23.3, df=1, 41, p<0.001) relative to comparison subjects (Figure 1). Some of these subjects had previously been studied for parvalbumin and/or somatostatin mRNA levels, mostly by in situ hybridization (see Table S1 in the online data supplement). In the newly studied schizophrenia subjects alone, we also found lower mRNA levels for parvalbumin (−26%; F=21.4, df=1, 22, p<0.001, N=23 pairs) and somatostatin (−30%; F=11.3, df=1, 18, p=0.004; N=19 pairs). However, across the entire cohort, not all schizophrenia subjects showed lower mRNA levels for parvalbumin (N=10) or somatostatin (N=8) relative to their matched comparison subjects (Figure 1).

a mRNA levels for schizophrenia subjects relative to matched comparison subjects in a pair are indicated by open circles. Data points to the right of the unity line indicate lower mRNA levels in the schizophrenia subject relative to the comparison subject and vice versa. Mean mRNA levels in schizophrenia subjects were statistically significantly lower for parvalbumin (mean=0.030, SD=0.010), somatostatin (mean=0.084, SD=0.042), and Lhx6 (mean=0.0098, SD=0.0021) and higher for calretinin (mean=0.027, SD=0.007) relative to comparison subjects (parvalbumin: mean=0.037, SD=0.007; somatostatin: mean=0.115, SD=0.029; Lhx6: mean=0.0107, SD=0.0013; calretinin: mean=0.025, SD=0.005). In contrast, Sox6 mRNA levels did not differ between schizophrenia subjects (mean=0.0111, SD=0.0018) and comparison subjects (mean=0.0105, SD=0.0016).
Calretinin mRNA levels were significantly elevated by 9% in schizophrenia subjects (F=4.6, df=1, 41, p=0.037) (Figure 1). Nineteen subject pairs were previously found to have no difference in calretinin mRNA expression (see Table S1 in the online data supplement) (7, 8, 28), and qPCR analysis of calretinin mRNA levels in these subject pairs similarly revealed no significant differences in schizophrenia (+2%; F=0.31, df=1, 18, p=0.58). However, calretinin mRNA levels were significantly higher in the new group of 23 schizophrenia subjects (+16%; F=6.7, df=1, 22, p=0.017).
Lhx6 and Sox6 mRNA Expression in Schizophrenia
qPCR analysis revealed that mean mRNA levels of Lhx6 (−10%; F=6.1, df=1, 40, p=0.018), but not Sox6 (F=2.5, df=1, 41, p=0.12), were lower in schizophrenia relative to comparison subjects (Figure 1). Consistent with their colocalization to the same interneuron subpopulations, Lhx6 and Sox6 mRNA levels were strongly correlated in comparison subjects (r=0.56, p<0.001; see Figure S3 in the online data supplement), but not in schizophrenia subjects (r=0.06, p=0.71), as expected for a disease effect on Lhx6 but not Sox6.
We next validated the finding of lower Lhx6 mRNA levels in schizophrenia using in situ hybridization in a limited number of subjects (i.e., the first 22 subject pairs in Table S1 [in the online data supplement] that had a sufficient number of available tissue sections). Mean Lhx6 mRNA levels were similarly lower when quantified by in situ hybridization (−13%; F=4.9, df=1, 21, p=0.038) (Figure 2A,B) or by qPCR (−12%) in these schizophrenia subjects. Furthermore, Lhx6 mRNA levels quantified by in situ hybridization and by qPCR were highly correlated in the same subjects (r=0.69, p<0.001; N=44 subjects).
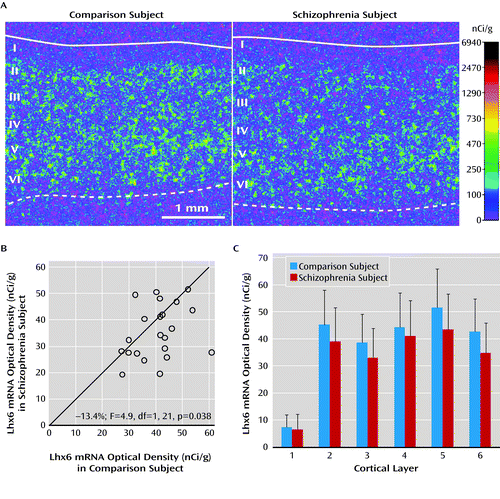
a In panel A, pseudocolored film autoradiographs of prefrontal cortical sections processed by in situ hybridization demonstrate lower Lhx6 mRNA levels in a schizophrenia subject relative to the matched comparison subject. The solid white line indicates the border between pia and layer 1, and the dashed line indicates the border between layer 6 and white matter. In panel B, open circles indicate average Lhx6 mRNA levels across gray matter of prefrontal cortical area 9 for schizophrenia subjects relative to matched comparison subjects in a pair. Data points to the right of the unity line indicate lower mRNA levels in the schizophrenia subject relative to the comparison subject and vice versa. Mean Lhx6 mRNA levels were 13% lower in schizophrenia subjects relative to matched comparison subjects. In panel C, laminar analysis of prefrontal cortical area 9 revealed that Lhx6 mRNA levels were lower by 14% in layer 2 (p=0.07), 15% in layer 3 (p<0.05), 7.4% in layer 4, 16% in layer 5 (p<0.05), and 18% in layer 6 (p<0.05) in schizophrenia subjects.
We then conducted a laminar analysis of Lhx6 mRNA levels to determine whether Lhx6 mRNA levels were lower in the cortical layers where parvalbumin neurons (layers 3–4) (7) and/or somatostatin neurons (layers 2, 5, and 6) (9) are most commonly found. Lhx6 mRNA levels were lower by 14% in layer 2 (F=3.7, df=1, 21, p=0.07), 15% in layer 3 (F=5.6, df=1, 21, p=0.028), 7.4% in layer 4 (F=0.94, df=1, 21, p=0.34), 16% in layer 5 (F=5.8, df=1, 21, p=0.025), and 18% in layer 6 (F=9.2, df=1, 21, p=0.005) (Figure 2C), reaching statistical significance in layers 3, 5, 6 and almost in layer 2.
We then used a grain counting analysis to determine whether Lhx6 mRNA expression is lower in all Lhx6-containing neurons and/or whether a subset of neurons is particularly affected (Figure 3). In schizophrenia subjects, the mean number of labeled neurons/mm2 was lower in layer 3 (−24%; F=10.6, df=1, 21, p=0.004) and layer 6 (−27%; F=9.1, df=1, 21, p=0.007) relative to comparison subjects. Furthermore, in schizophrenia subjects, mean grain density per labeled neuron was also lower in layer 3 (−17%; F=8.6, df=1, 21, p=0.008) and layer 6 (−19%; F=13.4, df=1, 21, p=0.001) relative to comparison subjects. These data suggest that a subset of neurons fail to express detectable levels of Lhx6 mRNA while the remaining neurons express lower Lhx6 mRNA levels in schizophrenia.
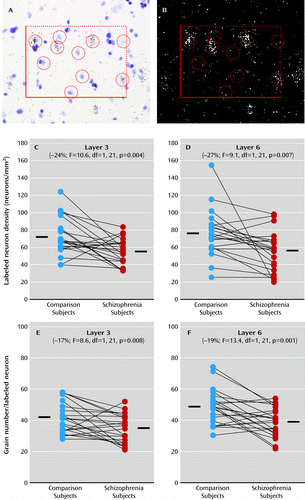
a Panel A is a representative brightfield image of a 120×170 μm sampling frame placed in layer 3 in which Nissl-stained neuronal nuclei were identified and sampled within inclusion and exclusion boundaries, indicated by broken and solid lines, respectively. Note that grain clusters identified in the darkfield image in panel B are located over some of the lightly Nissl-stained neuronal nuclei in the brightfield image but not over the darkly stained glial nuclei. Circles with a diameter of 22 μm were centered over all neuronal nuclei in every counting frame, and the number of grains in each circle was counted in the corresponding darkfield image. Panels C–F show cellular Lhx6 mRNA measurements in subjects with schizophrenia (red circles) and comparison subjects (blue circles) as expressed by labeled neurons/mm2 in layer 3 (panel C) and layer 6 (panel D) and by grains per positive neuron in layer 3 (panel E) and layer 6 (panel F). Subjects in each pair are connected by black lines, and the mean values for each subject group are indicated by a horizontal line. In schizophrenia subjects, the mean number of labeled neurons/mm2 was lower in layer 3 (mean=56.1, SD=14.8) and in layer 6 (mean=55.6, SD=22.7) relative to comparison subjects (mean=73.6, SD=21.6 and mean=75.8, SD=26.8, respectively). Furthermore, in schizophrenia subjects, the mean grain density per labeled neuron was also lower in layer 3 (mean=34.4, SD=9.1) and in layer 6 (mean=39.7, SD=9.2) relative to comparison subjects (mean=41.5, SD=9.9 and mean=49.0, SD=11.0, respectively).
Transcript Deficits in a Subset of Schizophrenia Subjects
We next determined whether lower mRNA levels for Lhx6, GAD67 (previously reported in this cohort [6]), parvalbumin, and somatostatin mRNAs were predominantly found in the same subset of schizophrenia subjects. Cluster analysis using standardized mRNA levels for Lhx6, GAD67, parvalbumin, and somatostatin for all schizophrenia and comparison subjects (N=84) yielded two distinct clusters. In one cluster (N=61 subjects), schizophrenia and comparison subjects were generally intermixed. However, the other cluster (N=23 subjects) was composed almost entirely of schizophrenia subjects (20/23 subjects) and contained 48% of all schizophrenia subjects (20/42) but only 7% of the comparison subjects (3/42) (Figure 4A). This “low-GABA-marker” subset of schizophrenia subjects (N=20) had lower levels of Lhx6 mRNA (F=29.1, df=2, 80, p<0.0001) relative to all other schizophrenia subjects (−26%; t=−7.06, p<0.0001; N=22) and comparison subjects (−22%; t=−6.55, p<0.0001; N=42); lower levels of GAD67 mRNA (F=48.4, df=2, 81, p<0.0001) relative to other schizophrenia subjects (−33%; t=−8.83, p<0.0001) and comparison subjects (−32%; t=−8.75, p<0.0001); lower levels of parvalbumin mRNA (F=20.3, df=2, 81, p<0.0001) relative to other schizophrenia subjects (−30%; t=−4.59, p<0.0001) and comparison subjects (−34%; t=−6.28, p<0.0001); lower levels of somatostatin mRNA (F=25.2, df=2, 80, p<0.0001) relative to other schizophrenia subjects (−39%; t=−4.56, p<0.0001) and comparison subjects (−46%; t=−7.06, p<0.0001); and no differences in calretinin mRNA (F=1.8, df=2, 81, p=0.17) or Sox6 mRNA (F=1.4, df=2, 81, p=0.26) (Figure 4B). In contrast, post hoc analyses revealed that mRNA levels for Lhx6, GAD67, parvalbumin, and somatostatin did not differ between the schizophrenia subjects not included in the low-GABA-marker cluster and all comparison subjects (Lhx6: t=1.57, p=0.12; GAD67: t=1.34, p=0.18; parvalbumin: t=−1.1, p=0.27; and somatostatin: t=−1.85, p=0.07).

a Panel A is a dendrogram illustrating a cluster analysis of standardized mRNA levels for storage time-adjusted Lhx6, GAD67, parvalbumin, and age-adjusted somatostatin across all schizophrenia and comparison subjects. In one cluster (left side), schizophrenia (orange) and comparison subjects (blue) were generally intermixed. However, the other cluster was composed almost entirely of schizophrenia subjects (red). The subset of schizophrenia subjects in panel B had lower average mRNA levels for Lhx6, GAD67, parvalbumin, and somatostatin, but not calretinin or Sox6, relative to all other schizophrenia subjects and to comparison subjects. Lhx6, GAD67, parvalbumin, and somatostatin mRNA levels did not differ between the remaining schizophrenia subjects and the comparison subjects.
*p<0.0001.
We next investigated whether the low-GABA-marker subset of schizophrenia subjects was more likely than the other schizophrenia subjects to have indicators of greater illness severity. Available demographic measures from psychological autopsy included factors predictive of illness severity such as male sex, diagnosis of schizophrenia rather than schizoaffective disorder, first-degree relative with schizophrenia, and earlier age at illness onset; and factors associated with greater illness severity, including suicide, no history of marriage, lower achieved socioeconomic status, and not living independently at time of death. However, no measure of greater illness severity was more common in the low-GABA-marker subset of schizophrenia subjects.
Effects of Substance Abuse and Psychotropic Medications
Schizophrenia is associated with elevated rates of substance abuse and long-term treatment with psychotropic medications. Consequently, we compared mRNA levels in schizophrenia subjects with and without a comorbid diagnosis of substance abuse or dependence or use of antipsychotic, antidepressant, or benzodiazepine medications at time of death. We found no relationship between any of these factors and mRNA levels for Lhx6 (see Figure S4 in the online data supplement), parvalbumin, or somatostatin, and these factors were not more common in the low-GABA-marker subset of schizophrenia subjects.
We also evaluated prefrontal Lhx6 mRNA levels in monkeys chronically exposed to haloperidol, olanzapine, or placebo (see Figure S5 in the online data supplement). (We previously reported no differences in mRNA levels for parvalbumin, somatostatin, and GAD67 in antipsychotic-exposed monkeys [2, 7, 9]). Lhx6 mRNA levels were slightly higher in haloperidol-exposed (+9%) and olanzapine-exposed monkeys (+6%) compared with placebo-exposed monkeys, although this difference did not achieve statistical significance (F=0.72, df=2, 10, p=0.51).
Discussion
In the prefrontal cortex of schizophrenia subjects, we found deficits in mRNA levels for Lhx6, an ontogenetic transcription factor selectively expressed by parvalbumin and somatostatin neurons in humans (24). Furthermore, our findings of lower mRNA levels for parvalbumin and somatostatin are remarkably similar to reports in other cohorts of schizophrenia subjects (5, 10). Interestingly, a subset of schizophrenia subjects was identified by the presence of consistent deficits in Lhx6, GAD67, parvalbumin, and somatostatin mRNAs. In contrast, not all GABA neuron markers were lower in this subset of schizophrenia subjects (e.g., calretinin), and not all mRNA species localized to parvalbumin and somatostatin neurons were deficient (e.g., Sox6). Taken together, these data suggest that deficient Lhx6 mRNA expression may contribute to alterations in phenotypic markers in prefrontal parvalbumin and somatostatin neurons in a subset of schizophrenia subjects.
Lhx6 is a transcription factor expressed by (future) parvalbumin and somatostatin neurons as they migrate from the medial ganglionic eminence to the cerebral cortex (11–14, 19, 20) as early as 7 weeks’ gestation in humans (16), and Lhx6 continues to be strongly expressed in this cell type-specific manner in adult human cortex (24). The pathogenetic consequences of low Lhx6 levels may depend on the developmental stage at which Lhx6 deficits first emerge in the disorder. For example, deficits in Lhx6 at the earliest gestational periods impair the migration of cortical parvalbumin and somatostatin neurons (19, 20). In schizophrenia, the densities of cortical Lhx6-positive neurons and somatostatin-positive neurons (9) are lower in the gray matter while the density of somatostatin-positive neurons is higher in the interstitial white matter (23); together, these findings are suggestive of arrested migration of some somatostatin neurons. Alternatively, a loss of Lhx6 that occurs after migration might interfere with the development of the GABA-ergic phenotype. For example, a lower density of cortical Lhx6-positive neurons could mean that these neurons are still present but have undetectable levels of Lhx6 mRNA. Consistent with this interpretation, the density of prefrontal parvalbumin mRNA-containing neurons was not altered in schizophrenia, but approximately 50% of these neurons lacked detectable GAD67 mRNA (7). As a third alternative, deficits in Lhx6 may appear in late adolescence/early adulthood after cellular maturation has been completed. In this case, the clustering of Lhx6, GAD67, parvalbumin, and somatostatin mRNA deficits in a subset of schizophrenia subjects may instead reflect the shared downstream consequences of some other upstream pathogenetic event that selectively affects parvalbumin and somatostatin neurons. However, our failure to detect a change in Sox6 mRNA, which is selectively expressed by cortical parvalbumin and somatostatin neurons (21, 22), is inconsistent with a general disease effect that affects all transcripts localized to these neurons. Finally, the absence of an effect of age on Lhx6 mRNA levels in schizophrenia suggests that lower Lhx6 mRNA levels are not attributable to a prolonged neurodegenerative process. Since our study was designed to detect alterations present in adulthood, we cannot directly determine the developmental stage at which deficits in Lhx6 mRNA levels, or any other GABA neuron markers, occur in the disorder. Interestingly, though, cognitive developmental delays have been reported in the premorbid stages of schizophrenia (32), even before age 1 (33). Thus, these lines of evidence suggest that the pattern of molecular abnormalities in prefrontal GABA neurons seen in a subset of schizophrenia subjects may reflect the long-lasting consequence of disturbances in the ontogeny of parvalbumin and somatostatin neurons that might have begun early in prenatal life.
The finding that deficits in certain GABA neuron-related mRNAs cluster together in a subset of schizophrenia subjects may help inform novel diagnostic strategies and individualized treatment approaches in schizophrenia. This subset of schizophrenia subjects with clear GABA neuron-related deficits may not necessarily have a more severe form of the illness, as suggested by the lack of difference in markers of illness severity from other schizophrenia subjects. However, one may predict that schizophrenia-related deficits in gamma band oscillations (34, 35), which are subserved by cortical parvalbumin neurons (36) and are important for prefrontal cortex-related cognitive processes (37), may be greater in the low-GABA-marker subset of schizophrenia subjects (38). Furthermore, approaches to clinically identifying schizophrenia patients with GABA neuron-related deficits might include positron emission tomography studies of shifts in extracellular GABA levels (39) or magnetic resonance spectroscopy (MRS) studies to quantify total cortical GABA levels. Interestingly, the evidence for two subsets of schizophrenia subjects, with or without robustly lower cortical GAD67 mRNA levels (Figure 4), suggests that inconsistencies in reports of cortical GABA levels by MRS (40–42) may reflect differences in the sampling of schizophrenia subjects (i.e., different studies may include a variable proportion of schizophrenia subjects with and without deficits in GABA neuron-related mRNAs). In addition, allelic variants or altered histone methylation of the promoter regions of GABA neuron-related genes, such as GAD1 (4, 43), that affect gene expression and have been associated with an increased risk for schizophrenia may be even more common in the low-GABA-marker subset of schizophrenia subjects. Finally, personalized medicine approaches, such as using GABA agents (44, 45) in schizophrenia patients with identified cortical GABA deficits, may increase the likelihood of a beneficial effect on cognitive functioning.
1 : Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 1995; 52:258–266Crossref, Medline, Google Scholar
2 : Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 2000; 57:237–245Crossref, Medline, Google Scholar
3 : Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 2000; 57:1061–1069Crossref, Medline, Google Scholar
4 : Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry 2007; 12:854–869Crossref, Medline, Google Scholar
5 : Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry 2010; 167:1479–1488Link, Google Scholar
6 : Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry 2011; 168:921–929Link, Google Scholar
7 : Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 2003; 23:6315–6326Crossref, Medline, Google Scholar
8 : Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry 2008; 165:479–489Link, Google Scholar
9 : Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex 2008; 18:1575–1587Crossref, Medline, Google Scholar
10 : Molecular determinants of dysregulated GABAergic gene expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 2009; 65:1006–1014Crossref, Medline, Google Scholar
11 : Origins of cortical interneuron subtypes. J Neurosci 2004; 24:2612–2622Crossref, Medline, Google Scholar
12 : The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron 2005; 48:591–604Crossref, Medline, Google Scholar
13 : Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb Cortex 2006; 16(Suppl 1):i82–i88Crossref, Medline, Google Scholar
14 : Selective depletion of molecularly defined cortical interneurons in human holoprosencephaly with severe striatal hypoplasia. Cereb Cortex 2009; 19:2196–2207Crossref, Medline, Google Scholar
15 : Interneurons in the developing human neocortex. Dev Neurobiol 2011; 71:18–33Crossref, Medline, Google Scholar
16 : Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb Cortex 2011; 21:1771–1782Crossref, Medline, Google Scholar
17 : Origin of GABAergic neurons in the human neocortex. Nature 2002; 417:645–649Crossref, Medline, Google Scholar
18 : Contributions of cortical subventricular zone to the development of the human cerebral cortex. J Comp Neurol 2005; 491:109–122Crossref, Medline, Google Scholar
19 : Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J Neurosci 2007; 27:3078–3089Crossref, Medline, Google Scholar
20 : Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J Comp Neurol 2008; 510:79–99Crossref, Medline, Google Scholar
21 : SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci 2009; 12:1238–1247Crossref, Medline, Google Scholar
22 : The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron 2009; 63:466–481Crossref, Medline, Google Scholar
23 : Increased interstitial white matter neuron density in the dorsolateral prefrontal cortex of people with schizophrenia. Biol Psychiatry 2011; 69:63–70Crossref, Medline, Google Scholar
24 : Selective expression of KCNS3 potassium channel α-subunit in parvalbumin-containing GABA neurons in the human prefrontal cortex. PLoS One 2012; 7(8):e43904 (doi:10.1371/journal.pone.0043904)Crossref, Google Scholar
25 : DSM-IV. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, D.C., American Psychiatric Association, 1994Google Scholar
26 : Alterations in metabotropic glutamate receptor 1α and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. Am J Psychiatry 2010; 167:1489–1498Link, Google Scholar
27 : Cortical opioid markers in schizophrenia and across postnatal development. Cereb Cortex 2012; 22:1215–1223Crossref, Medline, Google Scholar
28 : Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 2008; 13:147–161Crossref, Medline, Google Scholar
29 : Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3:34.1–34.11Crossref, Google Scholar
30 : Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch Gen Psychiatry 2008; 65:772–784Crossref, Medline, Google Scholar
31 : The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: a comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology 2005; 30:1649–1661Crossref, Medline, Google Scholar
32 : Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry 2010; 167:160–169Link, Google Scholar
33 : Early developmental milestones and risk of schizophrenia: a 45-year follow-up of the Copenhagen Perinatal Cohort. Schizophr Res 2010; 118:41–47Crossref, Medline, Google Scholar
34 : Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci USA 2006; 103:19878–19883Crossref, Medline, Google Scholar
35 : Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 2010; 35:2590–2599Crossref, Medline, Google Scholar
36 : Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009; 459:698–702Crossref, Medline, Google Scholar
37 : Gamma oscillations correlate with working memory load in humans. Cereb Cortex 2003; 13:1369–1374Crossref, Medline, Google Scholar
38 : Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6:312–324Crossref, Medline, Google Scholar
39 : Tiagabine increases [11C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology 2009; 34:624–633Crossref, Medline, Google Scholar
40 : Reduction of brain gamma-aminobutyric acid (GABA) concentrations in early-stage schizophrenia patients: 3T Proton MRS study. Schizophr Res 2009; 112:192–193Crossref, Medline, Google Scholar
41 : Elevated gamma-aminobutyric acid levels in chronic schizophrenia. Biol Psychiatry 2010; 68:667–670Crossref, Medline, Google Scholar
42 : GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J Neurosci 2010; 30:3777–3781Crossref, Medline, Google Scholar
43 : Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci 2007; 27:11254–11262Crossref, Medline, Google Scholar
44 : Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry 2008; 165:1585–1593Link, Google Scholar
45 : A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry 2011; 69:442–449Crossref, Medline, Google Scholar

