
A Genome Screen for Quantitative Trait Loci Influencing Schizophrenia and Neurocognitive Phenotypes
Abstract
Objective: Deficits in neurocognitive function have been demonstrated in individuals with schizophrenia and in the unaffected family members of these individuals. Genetic studies of such complementary traits, along with traditional analyses of diagnosis, may help to elucidate the biological pathways underlying familial liability to schizophrenia and related disorders. The authors conducted a multiplex, multigenerational family study using a genome-wide screen for schizophrenia and related neurocognitive phenotypes. Method: Participants were 1) 676 European American individuals from 43 families, ascertained through an individual with schizophrenia, and 2) 236 healthy comparison subjects. Participants were evaluated clinically and examined through the use of a computerized neurocognitive test battery that provided measures of accuracy and speed on the cognitive domains of abstraction and mental flexibility; attention; verbal, face, and spatial memory; language and reasoning; spatial and emotion processing; and sensorimotor dexterity. A genome-wide linkage screen was also performed. Healthy comparison subjects were included in order to obtain normative phenotypic data but were not genotyped. Results: Significant evidence for linkage of schizophrenia to chromosome 19q was observed. Analysis of cognitive traits revealed significant linkage to chromosome 5q for the domains of abstraction and mental flexibility. A variety of other neurocognitive traits also showed nominal evidence of linkage to the 5q region. Joint analyses with diagnosis suggested that this quantitative trait locus may also influence schizophrenia. Conclusions: Although chromosome 5 has been implicated in previous linkage studies of schizophrenia, the identification of the chromosome 19 quantitative trait locus is a novel finding. The identification of the chromosome 5 quantitative trait locus through linkage to neurocognitive phenotypes in the present study may inform functional hypotheses pertaining to how genotypes are connected to disease.
Cognitive deficits have been observed in individuals with schizophrenia and in the clinically unaffected relatives of these individuals, particularly in the domains of executive function, learning, and memory (1 – 7) . The appearance of cognitive deficits in schizophrenia patients and their unaffected relatives suggests that, rather than an outcome of the disease process, these deficits are part of the innate underlying distinct differences that make some individuals vulnerable to schizophrenia. Examining these complementary biological and behavioral phenotypes may be beneficial in genetic studies of a particular disorder and may provide valuable information about the pathophysiology that connects genotype to clinical disease (8) . Genetic studies of correlated quantitative phenotypes can complement traditional studies of disease outcome, since a subset of genes that influences a disorder may have larger genetic effects on a complementary trait than on the disease endpoint. These genes would be easier to detect at genome-wide significance levels in studies of a particular quantitative trait. Additionally, genetic analyses of complementary phenotypes can be used as a way to form functional hypotheses regarding how genotype connects to disease for genes that are identified through both diagnosis- and quantitative phenotype-based analyses.
In the present study, we conducted a genome-wide linkage screen of schizophrenia and measures of cognitive performance through the use of a computerized neurocognitive test battery that was designed to assess the following commonly affected cognitive domains in schizophrenia and related disorders: abstraction and mental flexibility; attention; verbal, face, and spatial memory; language and reasoning; spatial and emotion processing; and sensorimotor dexterity (9) . Efficiency of performance on each cognitive task was derived as the ratio of accuracy to speed of performance. For efficiency traits with significant or suggestive linkage signals, we explored whether these signals originated primarily from the 1) accuracy or 2) speed component of the efficiency phenotype by assessing linkage to the same chromosomal region for these two subphenotypes. These analyses were conducted in multiplex, multigenerational European American families that were ascertained through an individual with schizophrenia. As recently reported (9) , some aspects of neurocognition are significantly heritable in this sample. To our knowledge, the present study is the first genome-wide screen (either linkage or association) for genes that influence variation in several of these neurocognitive domains.
Method
Recruitment
The recruitment and study design have been described previously (9) , although the sample size of the present analyses is larger and consists of 676 individuals from 43 families, which is an average of 15.7 individuals examined per family. The families were ascertained through a European American individual with a DSM-IV diagnosis of schizophrenia who was required to have 1) at least one first-degree relative with schizophrenia or schizoaffective disorder, depressed type, and 2) an extended family of at least 10 first- and second-degree relatives who could be prospective study participants. All available first-, second-, and third-degree relatives ≥15 years of age were invited to participate in the study. In one family, the final best-estimate diagnosis of the secondary proband was schizoaffective disorder, bipolar type, which means that 42 families were multiplex and one was simplex according to study criteria. The simplex family was retained in the present analyses.
The study sample included 236 healthy European American individuals who were unrelated to the schizophrenia patients but were examined with the same clinical and neurocognitive instruments as the family members of the schizophrenia patients in order to provide baseline population data on the distribution of the quantitative phenotypes. Healthy comparison subjects were screened for axis I and axis II cluster A disorders and were psychiatrically, medically, and neurologically healthy, with no history of psychosis or mood disorders in their first-degree relatives. Although these unrelated individuals did not provide data regarding genetic linkage and were not genotyped, they were a valuable addition to the variance component-based genetic analyses utilized in the present study, since these analyses depend on estimating population parameters such as the mean and variance of each trait.
All participants provided informed consent, and the study was approved by the Institutional Review Board of each of the three participating institutions. In the case of minors <18 years old, assent was obtained from the child, and consent was obtained from a parent.
Phenotyping
Participants completed a computerized neurocognitive test battery designed to evaluate 1) abstraction and mental flexibility, 2) attention, 3) verbal memory, 4) face memory, 5) spatial memory, 6) language and reasoning, 7) spatial processing, 8) emotion processing, and 9) sensorimotor dexterity (10 , 11) . The emotion processing task included emotion intensity discrimination (9 , 12) and an identification task for the following emotions: happy, sad, angry, fearful, and neutral (13) . The computerized test battery automatically tabulated both the number of correct responses to the task (i.e., accuracy) and the median time (i.e., speed) for correct responses (in msec). From these two measures, we also calculated the ratio of accuracy to speed, which is a measure of efficiency. For sensorimotor dexterity, accuracy and efficiency were not analyzed, since >75% of participants had perfect scores, resulting in too little variation in sensorimotor dexterity traits within the study sample. Neurocognitive domain scores were transformed to their standard equivalents (z scores) based on the normative comparison sample. Further details regarding the individual tests of the computerized neurocognitive test battery and administration of the battery in the present sample are detailed elsewhere (9) .
DSM-IV clinical diagnoses were obtained through a standard consensus diagnostic process based on data from the Diagnostic Interview for Genetics Studies (version 2.0), which was administered to each participant; the Family Interview for Genetics Studies; and a review of any available medical records. Each case was reviewed by two investigators who were blind to the family relationships among the participants in order to arrive at a lifetime best-estimate final diagnosis for a particular subject. Best-estimate diagnoses for subjects with psychotic features and disagreement between the two raters were discussed in diagnostic meetings at each site, and particularly difficult cases were discussed among the investigators at the two ascertainment sites. Interrater reliability within and across sites was evaluated using videotaped interviews in order to maintain a kappa >0.8.
Among the 43 participating families, there were 106 individuals who had schizophrenia or schizoaffective disorder, depressed type. Most families had two affected members as a result of ascertainment of an affected relative pair. However, nine families had three affected members; two families had four affected members; and three families had five affected members. In addition, the following diagnoses were also present in family members: cluster A diagnoses (N=22), schizoaffective-bipolar disorder (N=4), delusional disorder (N=2), and brief psychotic disorder (N=1).
Of the participants with schizophrenia or schizoaffective disorder, depressed type, 75% were receiving treatment at the time of assessment. Some individuals were receiving multiple medications. These treatments included first-generation (typical) antipsychotics (33%), second-generation antipsychotics (48%), mood stabilizers (15%), and benzodiazepine (10%). Among the 25% of affected individuals who were not being treated at the time of assessment, approximately one-third had never been medicated. Additionally, one-third of the untreated individuals had discontinued treatment ≥6 months before assessment, and one-third had discontinued treatment within the 6 months prior to assessment. Effects of medication on neurocognitive measures have been examined extensively in previous studies and were found to be negligible or subtle (14 – 17) . Therefore, medication status was not considered in the present analyses.
Genotyping Autosomal Microsatellite Markers
DNA from immortalized cell lines was provided by the National Institute of Mental Health DNA repository at Rutgers University (New Brunswick, N.J.), and participants in the families were genotyped through the use of the ABI PRISM Linkage Mapping Set-MD10, Version 2.5 (Applied Biosystems, Foster City, Calif.) at the Southwest Foundation for Biomedical Research. This mapping set consists of 386 autosomal microsatellite markers selected from the Genethon human linkage map and designed to create a map with markers spaced approximately 10 cM apart.
Polymerase chain reactions were conducted through the use of True Allele Polymerase Chain Reactions Premix (Applied Biosystems, Foster City, Calif.), and amplification occurred according to the manufacturer’s specifications. After polymerase chain reactions were conducted, the products of separate reactions for each individual were pooled, and a labeled size standard was added to each pool. The pooled polymerase chain reaction products were loaded into an ABI PRISM 3100 Genetic Analyzer for laser-based automated genotyping. The autosomal microsatellite markers were detected and quantified by fluorescent emissions, and their sizes were estimated by comparison with the labeled size standard using the Genescan software package (Applied Biosystems, Foster City, Calif.). Genotypes were assigned using the Genotyper software package (Applied Biosystems, Foster City, Calif.). The average heterozygosity of the autosomal microsatellite markers was 79%.
Genotypic Data Cleaning
The program PREST was used to assess whether the reported pedigree relationships were consistent with the observed genotype data. Apparent inconsistencies were clarified with the data collection centers.
SimWalk2 was used to estimate error probabilities for each individual for each marker genotype. The mistyping probabilities generated by SimWalk2 were used to blank genotypes through the use of an iterative procedure until no further Mendelian inconsistencies remained. A total of 782 genotypes were blanked in order to resolve Mendelian inconsistencies, representing <0.5% of the total number of genotypes. SimWalk2 can also be used to detect genotyping errors that are consistent with Mendelian inheritance but result in the appearance of double recombinants within a statistically improbable chromosomal length. To eliminate erroneous genotypes that result in potential spurious double recombinants, genotypes with an error probability of >25% were blanked. This resulted in the blanking of 806 genotypes, representing <0.5% of the total genotypes.
Statistical Genetic Analyses
Maximum likelihood techniques, implemented in SOLAR, were used to estimate allelic frequencies for each marker, and the Loki program was used to compute multipoint identity-by-descent matrices through the use of marker map positions supplied with the ABI Prism Linkage Mapping Set. The Loki program uses Markov chain Monte Carlo methods to estimate the expected identity-by-descent sharing at a particular chromosomal location conditional on the genotypes at neighboring markers.
Linkage analyses were conducted using standard variance component methods as implemented in SOLAR (18) . Schizophrenia was analyzed using the liability threshold model (19 , 20) , a technique for analyzing categorical yes/no traits within a variance component framework. The liability threshold model utilizes data from both affected and unaffected individuals but does not require specification of a penetrance model. Linkage analyses with the liability threshold model are similar to affected and discordant sibling pair analyses in that the logarithm of the odds ratio (LOD) score is highest in regions where relatives who are concordant for diagnosis (both affected or both unaffected) share alleles that are identical by descent with each other but not with relatives with whom they are discordant. Individuals were considered 1) “affected” (N=106) if they had schizophrenia or schizoaffective disorder, depressed type; 2) “phenotype unknown” if they could not be assessed (N=28); or 3) “unaffected” (family individuals: N=542; healthy comparison subjects: 236) if they could not be classified as affected or phenotype unknown. Although some unexamined family members were suspected to be affected based on family report or medical records, individuals who were not directly interviewed as part of the present study were considered phenotype unknown. Age and sex terms were included in all analyses, and analyses of quantitative traits included conditioning the likelihood of the pedigree on the phenotype of the proband, an ascertainment correction (21) . Genome-wide p values were obtained through the use of methods described by Feingold et al. (22) , and simulations were used to estimate p values for the liability threshold model. In order to assess whether the loci detected through the cognitive phenotype also influenced schizophrenia, joint bivariate linkage analyses of diagnosis and neurocognitive traits were performed in regions that showed linkage to the quantitative traits.
Variance component linkage analyses are known to be sensitive to non-normality in trait distribution, with an inflation of type I error rate for leptokurtic trait distributions (23) . There are a variety of approaches for dealing with non-normality of trait distribution, and the optimal strategy may differ by trait (24) . Consequently, two methods were used in the present study to ensure proper type I error rate for each quantitative trait. First, heritability for each trait was estimated once with an inverse normal transformation and a second time with outliers >4 standard deviations from the mean removed and with the multivariate t distribution option specified in SOLAR. Second, the procedure that maximized the heritability was then used for linkage analyses, which were run only once for each trait. Most traits had heritabilities that were 0.01 to 0.10 points higher with normalization. Exceptions were spatial memory accuracy and face memory speed, both of which had slightly higher heritabilities under the multivariate t distribution and were analyzed using the t distribution model for linkage screening.
Results
Significant and suggestive LOD scores ≥1.9 on the genome-wide linkage screens for schizophrenia and efficiency measures for cognitive traits are detailed in Table 1 . Full results for the genome screen, with LOD scores every 10 cM on each chromosome for each trait as well as the map of genotyped markers, are provided in the data supplement accompanying the online version of this article. Two significant linkage signals, with genome-wide p values <0.05, were observed. Linkage analyses of schizophrenia resulted in a LOD score of 3.44 on chromosome 19q (genome-wide p=0.045 [ Figure 1 ]). This linkage signal extended over multiple pedigrees, with 10 families contributing 0.3–0.86 points to the overall LOD score. Of the cognitive traits, only analyses of efficiency of emotion discrimination resulted in a LOD score >1 in this region of chromosome 19 (LOD score=1.32 at 100 cM).

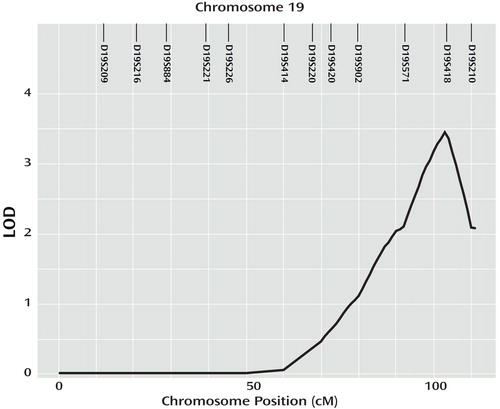
a Locations of genotyped markers are illustrated at the top of the plot.
The second significant linkage signal was a LOD score of 3.43 for efficiency of abstraction and mental flexibility on chromosome 5q (genome-wide p=0.011 [ Figure 2 ]). Consistent with the signal on chromosome 19, the chromosome 5 LOD score represented contributions from multiple pedigrees, with 12 families having individual LOD scores between 0.1 and 0.7. Accuracy, which was one of the component traits of abstraction and mental flexibility efficiency, also showed strong evidence for linkage to this region (LOD score=2.90). In the 25 cM centromeric to the abstraction and mental flexibility linkage signal, LOD scores within the 1.05–1.70 range were reported for the following domains: efficiency of attention, efficiency of verbal memory, verbal memory accuracy, language accuracy, and speed on the spatial processing test. Joint linkage analyses, with efficiency of abstraction and mental flexibility, and schizophrenia diagnosis at the location of the 5q linkage peak suggest that this quantitative trait locus may also influence schizophrenia (p=0.0024). The quantitative trait locus-specific correlation in this bivariate analysis was –0.28 and was significantly different from –1 (p=0.0025). The negative correlation was expected and indicates that individuals with a higher liability to schizophrenia had lower efficiencies of abstraction and mental flexibility, whereas those individuals with lower liability had higher efficiencies. However, if identical variants influenced both schizophrenia and efficiency of abstraction and mental flexibility, we would have expected a quantitative trait locus-specific genetic correlation of –1. The fact that the observed genetic correlation was significantly different from –1 implies a model of partial pleiotropy in which there were either multiple functional variants with overlapping but nonidentical influences on the two traits or gene-environment interactions that influenced one trait but not the other.

a Locations of genotyped markers are illustrated at the top of the plot.
For the suggestive linkage signals detailed in Table 1 , an examination of the LOD scores for the component traits of accuracy and speed showed that these signals were primarily driven by accuracy scores, which were generally slightly lower than the LOD scores for efficiency phenotype. The LOD score for verbal memory accuracy on chromosome 10 was 2.07. The LOD score for emotion processing fear accuracy on chromosome 11 was 2.47, and the LOD score for language and reasoning accuracy on chromosome 17 was 1.82. In the case of spatial processing, the LOD score for the accuracy phenotype on chromosome 20 was 2.41, which was higher than that for the efficiency ratio.
Discussion
In the present study, we reported genome-wide linkage screen results for eight efficiency traits of commonly affected domains of cognition in schizophrenia as well as analyses of schizophrenia diagnosis. Our findings obviously raise issues pertaining to multiple testing and how to interpret the significance of LOD scores. The fact that the neurocognitive phenotypes were highly correlated with each other and with schizophrenia adds to the complexity of this problem. The correlations among the quantitative traits ranged from 0.19–0.52, with all except three correlation pairs being >0.30. The fact that these traits were all correlated with each other rendered a straightforward Bonferroni correction for the number of genome-wide linkage screens (i.e., one per trait) inappropriately conservative. On the other hand, it was equally difficult to formally incorporate into the p values the increased confidence arising from the fact that there were multiple traits showing various levels of linkage to the chromosome 5q region. Consequently, our solution was to report the full extent of our analyses and the nominal support for each outcome and allow the reader to weigh these results in light of multiple testing and consistency across phenotypes.
Consistent with most complex trait linkage studies, the present study sample had power to detect only loci with moderate to large effects on a particular phenotype. An 80% power for a LOD score ≥3 was reached only for loci accounting for 20% of the residual trait variance after correction for the effects of age and sex. Of course, this was the power to detect a particular locus. Given that we expected multiple genes to influence each trait, our power to detect at least one of a group of loci was higher than our power to detect one specific locus. It should also be noted that linkage analyses are effective on the level of a chromosomal region, and therefore the detectable effect size in a linkage analysis represents the sum of all functional variants within a region. In order for a locus to account for at least 20% of the trait variance in a linkage, it is not necessary for any specific variant to have such a large effect but rather the sum of the effects of all the variants in a region must reach this threshold. Nevertheless, we were likely to have detected significant evidence of linkage only for a subset of loci with large effect sizes.
To our knowledge, this is the first study to report a quantitative trait locus influencing schizophrenia on chromosome 19q. It is possible that the liability threshold model used in our analyses might detect loci different from the more traditional affected sibling pair or penetrance model-based affected-only analyses. This is because our liability threshold model relied on both affected and unaffected individuals and evaluated both concordant and discordant pairs simultaneously. Thus, a signal detected by this model—but not by the models utilized in many affected sibling pair studies or older penetrance model-based studies—may represent a protective locus for which detection was driven by the unaffected individuals in the study sample.
Multiple linkage studies of schizophrenia have implicated chromosome 5q (25 – 31) , which is one of the regions with the most consistent evidence of linkage across studies as well as in a formal meta-analysis (32) . The gene neurogenin1 has been suggested as a possible candidate within this region and has been resequenced in 25 individuals from one linkage study (33) . No coding variants were identified in the study, although modest evidence of association (p=0.01–0.04) was observed with a single nucleotide polymorphism in the five prime untranslated region. However, since the sequencing was limited to the single 1,666 bp exon and 1,000 bp upstream and downstream, it is possible that unidentified functional variants may exist in more distal regulatory elements for the neurogenin1 gene that are further from the coding region.
Although the 5q region has been identified in previous studies, our observation of linkage with neurocognitive traits in the present study may advance our understanding of potential mechanisms connecting DNA variation in this region to risk for schizophrenia. The cluster of weakly significant LOD scores for a variety of traits near the significant linkage signal for abstraction and mental flexibility raises the possibility that this quantitative trait locus influences a broad range of cognitive abilities. These findings may be beneficial in fine mapping analysis. Furthermore, abstraction and mental flexibility represent a major deficit in schizophrenia, suggesting the involvement of frontal brain circuitry, including the dorsolateral and superior frontal regions, which are also implicated in schizophrenia (1) . The results of the present study encourage exploration of gene effects that could localize to frontal brain regions and increase the vulnerability of these regions to deficits in important executive functions.
The LOD score profiles on chromosome 5q for abstraction and mental flexibility and for verbal memory have two peaks ( Figure 2 ), which suggests that there might be more than one quantitative trait locus in this region influencing the traits of interest analyzed in the present study. In addition, fine mapping studies of diagnostic phenotype in schizophrenia have suggested multiple associations, with each conferring relatively modest risk (33) . This speculation is difficult to test formally at the level of the linkage analyses, but it is consistent with the finding of partial pleiotropy between the quantitative trait loci detected for abstraction and mental flexibility and schizophrenia and with the observation that some cognitive traits show both linkage peaks and some show only one peak. These findings would be expected in a case in which there were multiple quantitative trait loci, with some influencing all traits that show linkage to a region and some influencing only a subset of these traits. Additionally, many investigators have speculated that the linkage results that are most consistently replicated across studies and populations may be those for which there are multiple quantitative trait loci under the linkage peak, the effects of which are summed in linkage analyses of the region as a whole. A definitive resolution to this issue will likely have to await the identification of the specific gene or genes and functional variants underlying the linkages to the 5q region for abstraction and mental flexibility and schizophrenia.
1. Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM, Kester DB, Stafiniak P: Neuropsychological function in schizophrenia: selective impairment in memory and learning. Arch Gen Psychiatry 1991; 48:618–624Google Scholar
2. Elvevag B, Goldberg TE: Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol 2000; 14:1–21Google Scholar
3. Cannon TD, Zorrilla LE, Shtasel D, Gur RE, Gur RC, Marco EJ, Moberg P, Price RA: Neuropsychological functioning in siblings discordant for schizophrenia and healthy volunteers. Arch Gen Psychiatry 1994; 51:651–661Google Scholar
4. Faraone SV, Seidman LJ, Kremen WS, Pepple JR, Lyons MJ, Tsuang MT: Neuropsychological functioning among the nonpsychotic relatives of schizophrenic patients: a diagnostic efficiency analysis. J Abnorm Psychol 1995; 104:286–304Google Scholar
5. Egan MF, Goldberg TE, Gscheidle T, Weirich M, Rawlings R, Hyde TM, Bigelow L, Weinberger DR: Relative risk for cognitive impairments in siblings of patients with schizophrenia. Biol Psychiatry 2001; 50:98–107Google Scholar
6. Thompson JL, Watson JR, Steinhauer SR, Goldstein G, Pogue-Geile MF: Indicators of genetic liability to schizophrenia: a sibling study of neuropsychological performance. Schizophr Bull 2005; 31:85–96Google Scholar
7. Conklin HM, Curtis CE, Calkins ME, Iacono WG: Working memory functioning in schizophrenia patients and their first-degree relatives: cognitive functioning shedding light on etiology. Neuropsychologia 2005; 43:930–942Google Scholar
8. Gottesman II, Gould TD: The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003; 160:636–645Google Scholar
9. Gur RE, Nimgaonkar VL, Almasy L, Calkins ME, Ragland JD, Pogue-Geile MF, Kanes S, Blangero J, Gur RC: Neurocognitive endophenotypes in a multiplex multigenerational family study of schizophrenia. Am J Psychiatry 2007; 164:813–819Google Scholar
10. Gur RC, Ragland JD, Moberg PJ, Bilker WB, Kohler C, Siegel SJ, Gur RE: Computerized neurocognitive scanning, II: the profile of schizophrenia. Neuropsychopharmacology 2001; 25:777–788Google Scholar
11. Gur RC, Ragland JD, Moberg PJ, Turner TH, Bilker WB, Kohler C, Siegel SJ, Gur RE: Computerized neurocognitive scanning, I: methodology and validation in healthy people. Neuropsychopharmacology 2001; 25:766–776Google Scholar
12. Gur RE, Kohler CG, Ragland JD, Siegel SJ, Lesko K, Bilker WB, Gur RC: Flat affect in schizophrenia: relation to emotion processing and neurocognitive measures. Schizophr Bull 2006; 32:279–287Google Scholar
13. Kohler CG, Turner TH, Bilker WB, Brensinger CM, Siegel SJ, Kanes SJ, Gur RE, Gur RC: Facial emotion recognition in schizophrenia: intensity effects and error pattern. Am J Psychiatry 2003; 160:1768–1774Google Scholar
14. Saykin AJ, Shtasel DL, Gur RE, Kester DB, Mozley LH, Stafiniak P, Gur RC: Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch Gen Psychiatry 1994; 51:124–131Google Scholar
15. Censits DM, Ragland JD, Gur RC, Gur RE: Neuropsychological evidence supporting a neurodevelopmental model of schizophrenia: a longitudinal study. Schizophr Res 1997; 24:289–298Google Scholar
16. Goldberg TE, Goldman RS, Burdick KE, Malhotra AK, Lencz T, Patel RC, Woerner MG, Schooler NR, Kane JM, Robinson DG: Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: Is it a practice effect? Arch Gen Psychiatry 2007; 64:1115–1122Google Scholar
17. Keefe RS: Cognitive deficits in patients with schizophrenia: effects and treatment. J Clin Psychiatry 2007; 14(suppl 68):8–13Google Scholar
18. Almasy L, Blangero J: Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 1998; 62:1198–1211Google Scholar
19. Williams JT, Van Eerdewegh P, Almasy L, Blangero J: Joint multipoint linkage analysis of multivariate qualitative and quantitative traits, I: likelihood formulation and simulation results. Am J Hum Genet 1999; 65:1134–1147Google Scholar
20. Duggirala R, Williams JT, Williams-Blangero S, Blangero J: A variance component approach to dichotomous trait linkage analysis using a threshold model. Genet Epidemiol 1997; 14:987–992Google Scholar
21. Boehnke M, Lange K: Ascertainment and goodness of fit of variance component models for pedigree data. Prog Clin Biol Res 1984; 147:173–192Google Scholar
22. Feingold E, Brown PO, Siegmund D: Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent. Am J Hum Genet 1993; 53:234–251Google Scholar
23. Allison DB, Neale MC, Zannolli R, Schork NJ, Amos CI, Blangero J: Testing the robustness of the likelihood-ratio test in a variance-component quantitative-trait loci-mapping procedure. Am J Hum Genet 1999; 65:531–544Google Scholar
24. Blangero J, Williams JT, Almasy L: Robust LOD scores for variance component-based linkage analysis. Genet Epidemiol 2000; 1(suppl 19):S8–14Google Scholar
25. Silverman JM, Greenberg DA, Altstiel LD, Siever LJ, Mohs RC, Smith CJ, Zhou G, Hollander TE, Yang XP, Kedache M, Li G, Zaccario ML, Davis KL: Evidence of a locus for schizophrenia and related disorders on the short arm of chromosome 5 in a large pedigree. Am J Med Genet 1996; 67:162–171Google Scholar
26. Straub RE, MacLean CJ, O’Neill FA, Walsh D, Kendler KS: Support for a possible schizophrenia vulnerability locus in region 5q22–31 in Irish families. Mol Psychiatry 1997; 2:148–155Google Scholar
27. Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR, Crow TJ, Sherrington R, DeLisi LE: A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet 1998; 81:364–376Google Scholar
28. Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A, Petursson H, Curtis D: Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21–22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3–24 and 20q12.1–11.23. Am J Hum Genet 2001; 68:661–673Google Scholar
29. DeLisi LE, Mesen A, Rodriguez C, Bertheau A, LaPrade B, Llach M, Riondet S, Razi K, Relja M, Byerley W, Sherrington R: Genome-wide scan for linkage to schizophrenia in a Spanish-origin cohort from Costa Rica. Am J Med Genet 2002; 114:497–508Google Scholar
30. Devlin B, Bacanu SA, Roeder K, Reimherr F, Wender P, Galke B, Novasad D, Chu A, TCueno K, Tiobek S, Otto C, Byerley W: Genome-wide multipoint linkage analyses of multiplex schizophrenia pedigrees from the oceanic nation of Palau. Mol Psychiatry 2002; 7:689–694Google Scholar
31. Sklar P, Pato MT, Kirby A, Petryshen TL, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Verner A, Hudson TJ, Morley CP, Kennedy JL, Azevedo MH, Lander E, Daly MJ, Pato CN: Genome-wide scan in Portuguese island families identifies 5q31–5q35 as a susceptibility locus for schizophrenia and psychosis. Mol Psychiatry 2004; 9:213–218Google Scholar
32. Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HM, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, Sherrington R, O’Neill FA, Walsh D, Kendler KS, Ekelund J, Paunio T, Lonnqvist J, Peltonen L, O’Donovan MC, Owen MJ, Wildenauer DB, Maier W, Nestadt G, Blouin JL, Antonarakis SE, Mowry BJ, Silverman JM, Crowe RR, Cloninger CR, Tsuang MT, Malaspina D, Harkavy-Friedman JM, Svrakic DM, Bassett AS, Holcomb J, Kalsi G, McQuillin A, Brynjolfson J, Sigmundsson T, Petursson H, Jazin E, Zoega T, Helgason T: Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet 2003; 73:34–48Google Scholar
33. Fanous AH, Chen X, Wang X, Amdur RL, O’Neill FA, Walsh D, Kendler KS: Association between the 5q31.1 gene neurogenin1 and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 2007; 144:207–214Google Scholar

