
Schizophrenia Genetics and Dysbindin: A Corner Turned?
I keep the subject of my inquiry constantly before me, and wait till the first dawning opens gradually, by little and little, into a full and clear light.
—Isaac Newton (1)
Attempts to understand the genetic influences on schizophrenia go back to the roots of modern psychiatry. This effort has had four overlapping historical phases. The first confirmed the clinical impression of Kraepelin that schizophrenia “ran in families.” Uncontroversially, several generations of family studies of increasing methodologic sophistication verified that first-degree relatives of individuals with schizophrenia have an approximately 10-fold increased risk of illness. The second phase determined whether this familial aggregation was due to genetic or environmental factors. While more controversial, evidence from a series of adoption and twin studies showed that genetic factors contribute strongly to the etiology of schizophrenia.
These first phases assessed the aggregate effects of genes averaged over the genome. By contrast, the third or “gene localization” phase has used linkage to attempt to localize susceptibility genes to specific chromosomal regions. While findings from the first two phases of schizophrenia genetics were marked by substantial cross-study consistency, linkage studies of schizophrenia produced inconsistent results and poor rates of replication. Two findings suggest that, despite methodologic difficulties, linkage analysis can produce replicable results in schizophrenia. First, random a priori subsets of the Irish Study of High-Density Schizophrenia Families produced replicable evidence for linkage in three of four major genomic regions (2). More important, a careful meta-analysis of 20 schizophrenia genome scans produced evidence that these studies agreed, much more than would be expected by chance, on those regions that likely contained susceptibility genes (3).
I outline here recent exciting developments in the fourth phase of schizophrenia genetics, which is attempting to identify specific susceptibility genes. This story begins in 1995 when, during the first stages of analysis of the Irish Study of High-Density Schizophrenia Families, linkage was detected on chromosome 6p24-22 (4). Evidence for linkage in this region, which was replicated by some but not other groups (5, 6), was in aggregate sufficient to initiate a fine mapping project in the Irish sample under one part of this broad linkage peak. Family-based analyses produced rather clear evidence for an association between schizophrenia and a set of DNA markers in the gene DTNBP1 (dystrobrevin binding protein 1) or dysbindin-1 (7). Dysbindin-1, a 40–50-kDa protein that is expressed in neurons in many areas of the mouse and human brain, is named for its capacity to bind dystrobrevins (8, 9), proteins that are part of the dystrophin glycoprotein complex (10).
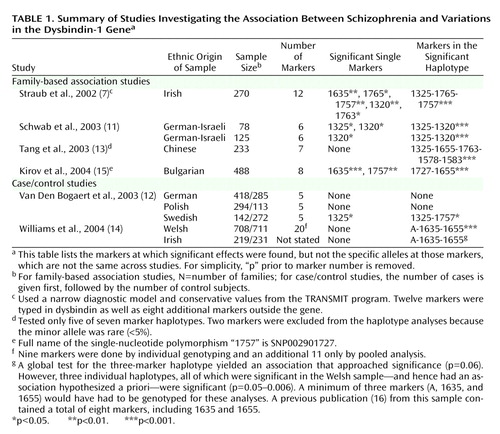
Since publication of this report in July 2002, replication efforts have been published from nine samples in five reports (11–15) (Table 1). (In addition, one negative study [16] was superseded by a positive report [14] with typing of additional markers, and further analyses in the Irish Study of High-Density Schizophrenia Families defined more clearly a high-risk haplotype [17].) Table 1 reports results for individual markers and for haplotypes. Haplotypes are distinctive blocks of DNA, identified by a particular set of alleles at a group of markers, that are so small that they are nearly always inherited together as a unit. (A marker specifies a location on the DNA molecule, while the allele indicates the particular base pair at that location.) Generally, haplotypes provide more power for association studies than do single markers.
Using single markers and haplotypes, significant associations were found in, respectively, four of nine and seven of nine of the replication samples. Between-study variation was found both in the significant single markers and the most significant haplotype. In addition, the alleles at the markers that were associated with schizophrenia were not identical across samples.
We have little experience in the interpretation of such data. Each study had a clear a priori prediction: markers or haplotypes in the dysbindin gene would be associated with schizophrenia. However, multiple tests were made of individual markers and haplotypes. Publication bias cannot be ruled out—although other positive reports have been presented in abstract form (18). The most important question we can ask is, given these findings, what is the probability that the hypothesis first put forward in the original report (4)—that variation in the dysbindin gene influences risk for schizophrenia—is false? No definitive answer to this critical question is yet possible. Prudence is suggested given the poor prior track record of claims for single gene effects in psychiatric illness. However, in aggregate, the results are strongly suggestive and argue that it is unlikely, and perhaps highly unlikely, that the original report was in error.
A secondary but still important question: if dysbindin is a susceptibility gene for schizophrenia, what can we say about its effect size? Not counting the original report (where the effect size might be biased upward), odds ratios reported or calculated from replication reports were 1.87 (marker 1325 combined across both samples [11]), 1.70 (marker 1325 in the one positive sample [12]), 1.23 (high-risk haplotype [13]), 1.40 (high-risk haplotype combined across both samples [14]), and 1.58 (marker 1635 [15]). If dysbindin is a susceptibility gene for schizophrenia, it is likely to be of small to moderate effect and account for a modest proportion of total genetic risk.
Very recently, two postmortem studies have added intriguing new information to the dysbindin-schizophrenia story. Talbot et al. (18) studied postmortem brains of two sets of matched pairs of schizophrenia and normal comparison individuals (N=17 and N=15, respectively). Using immunohistochemical methods, they found, in both samples, significant reductions of presynaptic dysbindin-1 in the hippocampal formation of the schizophrenia subjects, in particular in the terminal fields of intrinsic, glutamatergic afferents of the subiculum, the hippocampus proper, and the inner molecular layer of the dentate gyrus. These results were unlikely to result from neuroleptic exposure. Weickert et al. (19) studied levels of dysbindin messenger RNA in postmortem brains for 15 normal subjects and 14 individuals with schizophrenia. Dysbindin mRNA was widespread in the brain and was significantly reduced in multiple layers of the dorsolateral prefrontal cortex in schizophrenia subjects. This reduction was specific and not seen with other markers of synaptic integrity. They also reported that dysbindin mRNA levels were significantly correlated with four of 11 markers in the dysbindin gene.
In aggregate, these results are exciting, but it is important to emphasize what we do not know about the schizophrenia-dysbindin connection. We have no clear molecular “smoking gun”—a DNA variant that is both associated with schizophrenia and produces a distinct change in dysbindin structure or function. We have only the dimmest idea of how variation in dysbindin might contribute to the pathophysiology of schizophrenia. Given that only a subset of individuals with schizophrenia carry the high-risk dysbindin haplotype, why were reduced levels of dysbindin seen in the hippocampus of nearly all affected cases (18)? We do not have a single high-risk haplotype, and it remains unclear how to understand differences across samples. While multiple pathogenic mutations in dysbindin could explain these findings, it is perplexing to have such divergent results from ethnically similar populations.
So perhaps a corner has been turned in our long struggle to understand the genetic basis of schizophrenia. Furthermore, dysbindin is not the only gene where replicated evidence for association is emerging (see reference 6 for a recent review). For the genetics of schizophrenia, it may no longer be outlandish to hope that we will see “the first dawning [which] opens gradually, by little and little, into a full and clear light.”
 |
From the Virginia Institute for Psychiatric and Behavioral Genetics and the Departments of Psychiatry and Human Genetics, Medical College of Virginia of Virginia Commonwealth University, Richmond, VA. Address reprint requests to Dr. Kendler, Department of Psychiatry, P.O. Box 980126, Richmond, VA 23298-0126. Many individuals contributed critically to the development of the Irish Study of High-Density Schizophrenia Families and the subsequent work leading to the discovery of dysbindin, prominent among whom are Dermot Walsh, M.B., Tony O’Neill, M.D., Charles MacLean, Ph.D., and Richard Straub, Ph.D. The work, done in collaboration with the Health Research Board, Dublin, Ireland, and The Queen’s University, Belfast, Northern Ireland, was supported by NIMH grants MH-41953, MH-52537, and MH-45390. Brien Riley, Ph.D., Alan Gruenberg, M.D., and Douglas Levinson, M.D., provided comments on an earlier version of this essay.
1. http://www.quotationspage.com/quotes/Isaac_Newton/Google Scholar
2. Straub RE, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, O’Neill FA, Walsh D, Kendler KS: Genome-wide scans of three independent sets of 90 Irish multiplex schizophrenia families and follow-up of selected regions in all families provides evidence for multiple susceptibility genes. Mol Psychiatry 2002; 7:542–559Crossref, Medline, Google Scholar
3. Lewis CM, Levinson DF, Wise LH, Delisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HM, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, Sherrington R, O’Neill FA, Walsh D, Kendler KS, Ekelund J, Paunio T, Lonnqvist J, Peltonen L, O’Donovan MC, Owen MJ, Wildenauer DB, Maier W, Nestadt G, Blouin JL, Antonarakis SE, Mowry BJ, Silverman JM, Crowe RR, Cloninger CR, Tsuang MT, Malaspina D, Harkavy-Friedman JM, Svrakic DM, Bassett AS, Holcomb J, Kalsi G, McQuillin A, Brynjolfson J, Sigmundsson T, Petursson H, Jazin E, Zoega T, Helgason T: Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet 2003; 73:34–48Crossref, Medline, Google Scholar
4. Straub RE, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D, Kendler KS: A potential vulnerability locus for schizophrenia on chromosome 6p24–22: evidence for genetic heterogeneity. Nat Genet 1995; 11:287–293Crossref, Medline, Google Scholar
5. Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8: Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Am J Med Genet Neuropsychiatr Genet 1996; 67:580–594Crossref, Medline, Google Scholar
6. Owen MJ, Williams NM, O’Donovan MC: The molecular genetics of schizophrenia: new findings promise new insights. Mol Psychiatry 2004; 9:14–27Crossref, Medline, Google Scholar
7. Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’Neill FA, Walsh D, Kendler KS: Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 2002; 71:337–348Crossref, Medline, Google Scholar
8. Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ: Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem 2001; 276:24232–24241Crossref, Medline, Google Scholar
9. Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell’Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT: Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 2003; 35:84–89Crossref, Medline, Google Scholar
10. Blake DJ, Weir A, Newey SE, Davies KE: Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 2002; 82:291–329Crossref, Medline, Google Scholar
11. Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, Lerer B, Rietschel M, Trixler M, Maier W, Wildenauer DB: Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet 2003; 72:185–190Crossref, Medline, Google Scholar
12. Van Den Bogaert A, Schumacher J, Schulze TG, Otte AC, Ohlraun S, Kovalenko S, Becker T, Freudenberg J, Jonsson EG, Mattila-Evenden M, Sedvall GC, Czerski PM, Kapelski P, Hauser J, Maier W, Rietschel M, Propping P, Nothen MM, Cichon S: The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am J Hum Genet 2003; 73:1438–1443Crossref, Medline, Google Scholar
13. Tang JX, Zhou J, Fan JB, Li XW, Shi YY, Gu NF, Feng GY, Xing YL, Shi JG, He L: Family-based association study of DTNBP1 in 6p22.3 and schizophrenia. Mol Psychiatry 2003; 8:717–718Crossref, Medline, Google Scholar
14. Williams NM, Preece A, Morris DW, Spurlock G, Bray NJ, Stephens M, Norton N, Williams H, Clement M, Dwyer S, Curran C, Wilkinson J, Moskvina V, Waddington JL, Gill M, Corvin AP, Zammit S, Kirov G, Owen MJ, O’Donovan MC: Identification in 2 independent samples of a novel schizophrenia risk haplotype of the dystrobrevin binding protein gene (DTNBP1). Arch Gen Psychiatry 2004; 61:336–344Crossref, Medline, Google Scholar
15. Kirov G, Ivanov D, Williams NM, Preece A, Nikolov I, Milev R, Koleva S, Dimitrova A, Toncheva D, O’Donovan MC, Owen MJ: Strong evidence for association between the dystrobrevin binding protein 1 gene (DTNBP1) and schizophrenia in 488 parent-offspring trios from Bulgaria. Biol Psychiatry 2004; 55:971–975Crossref, Medline, Google Scholar
16. Morris DW, McGhee KA, Schwaiger S, Scully P, Quinn J, Meagher D, Waddington JL, Gill M, Corvin AP: No evidence for association of the dysbindin gene [DTNBP1] with schizophrenia in an Irish population-based study. Schizophr Res 2003; 60:167–172Crossref, Medline, Google Scholar
17. van den Oord EJ, Sullivan PF, Jiang Y, Walsh D, O’Neill FA, Kendler KS, Riley BP: Identification of a high-risk haplotype for the dystrobrevin binding protein 1 (DTNBP1) gene in the Irish Study of High-Density Schizophrenia Families. Mol Psychiatry 2003; 8:499–510Crossref, Medline, Google Scholar
18. Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, Hahn CG, Siegel SJ, Trojanowski JQ, Gur RE, Blake DJ, Arnold SE: Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest 2004; 113:1353–1363Crossref, Medline, Google Scholar
19. Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR, Kleinman JE: Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry 2004; 61:544–555Crossref, Medline, Google Scholar

