
Low Muscarinic Receptor Binding in Prefrontal Cortex From Subjects With Schizophrenia: A Study of Brodmann’s Areas 8, 9, 10, and 46 and the Effects of Neuroleptic Drug Treatment
Abstract
OBJECTIVE: Aberrant cholinergic inputs and synaptic neurotransmission in the prefrontal cortex induce cognitive impairment, which is a central feature of schizophrenia. Postsynaptic excitatory muscarinic cholinergic M1 and M4 receptors are the major cholinoceptive targets in the prefrontal cortex and hence may be involved in the pathology and/or pharmacotherapeutics of schizophrenia. METHOD: Using quantitative autoradiography, the authors analyzed the binding of the M1/M4 receptor selective antagonist [3H]pirenzepine in prefrontal cortex (Brodmann’s areas 8, 9, 10, and 46) from schizophrenia patients who had (N=6) or had not (N=11) been treated with the anticholinergic agent benztropine mesylate and from normal comparison subjects (N=20). Moreover, preliminary studies of [3H]pirenzepine binding in rat frontal cortex following administration of antipsychotic drugs or benztropine mesylate were performed. RESULTS: Relative to those of comparison subjects, the mean levels of [3H]pirenzepine binding were significantly lower in Brodmann’s areas 9 and 46 of the schizophrenia patients not treated with benztropine mesylate (18% lower in Brodmann’s area 9 and 21% lower in Brodmann’s area 46) and in all four examined regions of the patients who had received benztropine (51%–64% lower). Antipsychotic or anticholinergic drugs tended to increase or have no effect on the density of [3H]pirenzepine-labeled receptors in rat frontal cortex. CONCLUSIONS: Because M1 and M4 receptors are critical to the functions of prefrontal cortical acetylcholine, the present findings suggest a functional impairment in cholinergic neurotransmission in schizophrenia and the possibility that muscarinic receptors are involved in the pharmacotherapeutics of the disorder.
Several lines of evidence suggest that the prefrontal cortex, including Brodmann’s areas 8 to 10 and 44 to 47 (1, 2), is important to the pathology and treatment of schizophrenia. Neuropathological findings include low dendritic spine density on pyramidal neurons (3) and low levels of synaptophysin (a presynaptic terminal protein) (4–6) in the prefrontal cortex of schizophrenia patients. In addition, patients with schizophrenia perform poorly on cognitive tasks, especially those involving short-term memory and attention, which are subserved by neural mechanisms intrinsic to prefrontal cortex circuitry (7, 8).
Cholinergic afferents from the nucleus basalis of Meynert project to all layers of the cerebral cortex (9), account for 70%–80% of cortical cholinergic input (10–12), constitute a major regulatory pathway of prefrontal cortical functioning (9, 13, 14), and are important to higher cognitive processes (12, 15). Specifically, cortical cholinergic inputs are hypothesized to mediate an individual’s ability to detect, select, and process sensory or associational information essential to cognition (12). Lesions in the nucleus basalis of Meynert induce cortical cholinergic denervation (16); decrease baseline levels of acetylcholine in the frontal cortex (17); increase frontal cortical muscarinic cholinergic receptor levels, sensitivity, and mRNA (18–21); and impair learning and memory (22, 23). Accordingly, dysfunction of cholinergic input from the nucleus basalis of Meynert to the prefrontal cortex may primarily or secondarily contribute to aberrant prefrontal cortical activation and the emergence of cognitive symptoms associated with schizophrenia and other neuropsychiatric disorders.
Because postsynaptic excitatory muscarinic cholinergic M1 and M4 receptors are the major cholinoceptive targets in the prefrontal cortex (24), and hence essential to the circuitry between the nucleus basalis of Meynert and prefrontal cortex, we hypothesize that M1 and M4 receptors are primarily or secondarily altered in schizophrenia (25). Moreover, on the basis of the putative high-affinity, antimuscarinic actions of typical and atypical antipsychotic drugs, such as thioridazine, clozapine, and olanzapine (26–29), and the commonly administered anticholinergic benztropine mesylate (used to treat extrapyramidal side effects [30, 31]), we propose that M1 and M4 receptors are involved in the pharmacotherapeutics of schizophrenia. Therefore, using quantitative autoradiography, we measured the specific binding of the M1/M4 selective receptor antagonist [3H]pirenzepine (25, 32) in prefrontal cortex (Brodmann’s areas 8, 9, 10, and 46) from schizophrenia patients not treated or treated with benztropine and normal comparison subjects. Moreover, we performed preliminary studies of specific [3H]pirenzepine binding in frontal cortex from rats given or not given antipsychotic drugs or benztropine.
Method
Human Tissue Studies
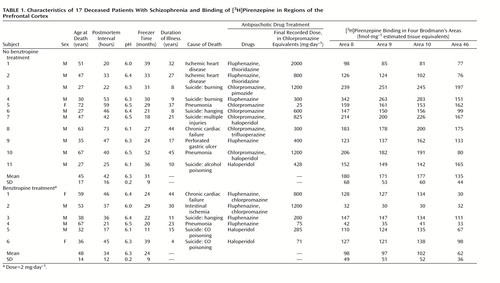
Postmortem human brain tissue was obtained during autopsy at the Victorian Institute of Forensic Medicine. The Human Ethics Committee of the Mental Health Research Institute of Victoria provided ethical approval for the collection of tissue. Specifically, prefrontal cortex (Brodmann’s areas 8, 9, 10, and 46) was collected from the left brain hemispheres of 17 patients with a diagnosis of schizophrenia and 20 subjects with no clinical history of psychiatric illness, neuroleptic exposure, or histopathological evidence of neurological disease (comparison subjects). Using a diagnostic instrument for brain studies (33), a senior psychiatrist and psychologist extensively reviewed the case histories of subjects with schizophrenia and confirmed the diagnosis according to DSM-III-R criteria. For each subject with schizophrenia, the duration of illness (the time from first hospital admission to death), final recorded antipsychotic drug dose (converted to chlorpromazine equivalents [34]), and use of benztropine was determined (Table 1).
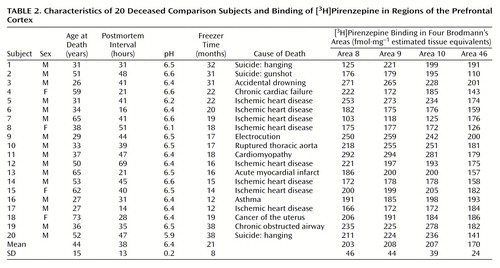
The age, postmortem interval, and sex of the subjects studied were determined (Table 1 and Table 2). The postmortem interval was taken as the time between witnessed death and autopsy. In cases where the death was not witnessed, tissue was collected only from subjects who had been seen alive within 5 hours of being found dead. In this instance, the postmortem interval was taken as the period starting half way between the last sighting of the subject while still alive and the time when he or she was found dead and ending at autopsy. In all cases, the cadavers were stored at 4°C within 5 hours of being discovered. After collection, the tissue was stored at –70°C until required (freezer time) (Table 1 and Table 2). To control for the effects of agonal state on the tissue collected, the pH of the brain tissue was determined as described previously (35).
Serial sections (four 20-μm sections per subject) of Brodmann’s areas 8, 9, 10, and 46 were coronally cut at –20°C by using a cryomicrotome, thaw-mounted onto chrome-alum/gelatin-coated glass slides, and stored at –70°C until required. Selection of tissue blocks was standardized by using a set of standard landmarks (36). Brodmann’s area 8 was taken as occupying the region posterior to Brodmann’s area 9 and anterior to Brodmann’s area 6, including the superior and middle frontal gyri immediately anterior to the superior precentral sulcus. Brodmann’s area 8 is wide on the superior surface, extending from the callosomarginal sulcus on the medial surface and narrowing laterally to the middle frontal gyrus. Brodmann’s area 9 was taken as being posterior to Brodmann’s area 10 and including the superior and middle frontal gyri. The medial surface of Brodmann’s area 9 was defined by the callosomarginal gyrus, while laterally it stopped ventrally in the region of the inferior frontal sulcus. Brodmann’s area 10 included the anterior quarter of the superior and middle frontal gyri on the convexity of the hemisphere, not extending medially as far as the callosomarginal gyrus. Inferomedially, Brodmann’s area 10 was demarcated by the superior rostral sulcus. Finally, Brodmann’s area 46 included the middle third of the middle and the anterior part of the inferior frontal gyri at the transition to the orbital surface.
Rat Tissue Studies
For the antipsychotic drug studies, 6-week-old male Sprague-Dawley rats (100–150 g, N=3 per group) were treated orally for 1 month (short-term) or 3 months (long-term) with vehicle (H2O; comparison rats), haloperidol, clozapine, chlorpromazine, or thioridazine. The haloperidol was initially dissolved in a minimal volume of 10% acetic acid (<0.001% final) and diluted with distilled water. Clozapine, chlorpromazine, and thioridazine were similarly dissolved in 0.1M HCl (<0.02 nM final), distilled water, and 10% ethyl alcohol (<0.001% final), respectively, and diluted with distilled water.
For the benztropine studies, 6-week-old male Sprague-Dawley rats (100–150 g, N=3 per group) were treated for 1 month with vehicle or benztropine. The benztropine was dissolved in distilled water and administered orally at a dose of 0.1 mg·kg–1·day–1(37).
To ensure that each rat received an appropriate amount of drug, the amount of each drug in drinking water was modified according to the measured daily water intake and weekly body weight of the subjects. At the completion of drug administration, the rats received drug-free drinking water for 72 hours before being killed by decapitation. The brains were rapidly removed, frozen in isopentane on dry ice, and stored at –70°C. Serial sections of rat frontal cortex (six 20-μm sections per subject) were coronally cut (so as to correspond to plates 5–7 from Paxinos and Watson [38]) at –20°C by using a cryomicrotome, thaw-mounted onto chrome-alum/gelatin-coated glass slides, and stored at –70°C until required.
In Situ [3H]Pirenzepine Binding With Autoradiography
The in vitro [3H]pirenzepine binding studies with autoradiography were performed as previously described (25). Briefly, mounted tissue sections were incubated with 15 nM [3H]pirenzepine (specific activity: 72 Ci/mmol) (New England Nuclear Life Science Products, Boston) in the absence (total binding; human studies: two sections, rat studies: three sections) or presence (nonspecific binding; human studies: two sections, rat studies: three sections) of 1 μM quinuclidinyl xanthene-9-carboxylate hemioxilate tetraoxilate (Research Biochemicals International, Natick, Mass.). The sections were washed, air dried, and apposed to Amersham [3H]-Hyperfilm (Amersham Pharmacia Biotech UK Ltd., Little Chalfont, Buckinghamshire, U.K.) with Amersham 3H micro-scales (Amersham Pharmacia Biotech UK Ltd., Little Chalfont, Buckinghamshire, U.K.) for up to 5 weeks. The images were analyzed by using a microcomputer imaging device and image analysis system (Imaging Research, Inc., St. Catherine’s, Ont., Canada). The results are expressed as femtomoles per milligram of estimated tissue equivalents. Specific radioligand binding was calculated as total binding minus nonspecific binding.
Statistical Analysis
One-way analyses of variance with least significant difference post hoc tests were used to compare the mean radioligand binding, age, postmortem interval, freezer time, and pH of the schizophrenia patients treated with benztropine, the untreated patients, and the comparison subjects, as well as levels of receptor binding in the frontal cortex of the rats given antipsychotic drugs and comparison rats for each length of administration. The relationships between receptor binding in human prefrontal cortex and subject age, postmortem interval, freezer time, pH, and, where relevant, duration of illness and final recorded antipsychotic drug dose were assessed by using Pearson product moment correlation coefficients, derived by using an assumed straight-line fit. A one-way analysis of covariance (ANCOVA) with least significant difference post hoc tests were used to determine whether freezer time was a confounding variable influencing the apparent differences between radioligand binding in the different groups. An unpaired t test was used to compare the mean levels of receptor binding in the frontal cortices of the rats given benztropine and the comparison rats.
Results
Human Tissue Studies
Individual levels of [3H]pirenzepine binding are shown in Table 1 and Table 2. The top four panels of Figure 1 contain sample images of [3H]pirenzepine binding (specific binding greater than 95% of total binding) in the prefrontal cortex of human subjects. The mean levels were significantly lower in Brodmann’s areas 9 (18% lower) and 46 (21% lower) of the prefrontal cortex from the schizophrenia patients not treated with benztropine than in the comparison subjects (Figure 2). Nonsignificant differences were seen in Brodmann’s areas 8 (11% lower) and 10 (15% lower). For the schizophrenia patients who were treated with benztropine, the mean levels of [3H]pirenzepine binding were significantly lower in all regions studied (52%, 53%, 51%, and 64% lower in Brodmann’s areas 8, 9, 10, and 46, respectively) than in the schizophrenia patients not treated with benztropine (Figure 2).
The mean age, postmortem interval, and tissue pH did not differ between the groups studied (Table 1 and Table 2). While the mean freezer time for tissue from the comparison group was significantly shorter than that for the schizophrenia group not treated with benztropine (F=4.31, df=2, 34, p=0.006), no other group differences were found. Radioligand binding in the prefrontal cortex from the groups studied did not correlate with age, postmortem interval, freezer time, or pH. ANCOVA showed that there was no significant effect of freezer time on the comparison of [3H]pirenzepine binding in the tissue from the groups studied (Figure 2). Finally, correlation analyses did not show a relationship between [3H]pirenzepine binding in the schizophrenia patients and the final recorded antipsychotic drug dose or duration of illness.
Rat Tissue Studies
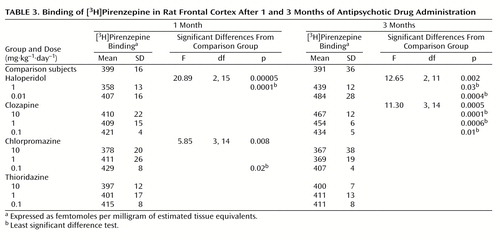
The bottom of Figure 1 represents receptor-bound [3H]pirenzepine levels in rat frontal cortex (specific binding greater than 95% of total binding). The effects of antipsychotic drug administration varied from one drug to another (Table 3). Although the rats that received short-term administration of 0.01 mg·kg–1·day–1 of haloperidol showed no difference in [3H]pirenzepine binding from the comparison rats, 1 mg·kg–1·day–1 produced significantly lower (10%) [3H]pirenzepine binding. In contrast, long-term treatment with either 0.01 or 1 mg·kg–1·day–1 of haloperidol caused significantly higher receptor-bound [3H]pirenzepine levels (24% and 12%, respectively) than those in the comparison rats. Short-term administration of 0.1, 1, or 10 mg·kg–1·day–1 of clozapine had no effect on [3H]pirenzepine binding. However, as with haloperidol, all doses of clozapine produced significantly higher levels (11%–19%) of labeled receptors after long-term drug administration. While short-term administration of 0.1 mg·kg–1·day–1 of chlorpromazine induced 8% higher [3H]pirenzepine binding than that in the comparison rats, no significant effects of other chlorpromazine treatments were found. Finally, no significant difference from the comparison value was measured after thioridazine administration at any dose for either period of time.
There was no difference between the rats given only vehicle and the rats that received 0.1 mg·kg–1·day–1 of benztropine for 1 month in [3H]pirenzepine-bound receptor levels (mean=387, SD=20, versus mean=387, SD=35) (t=0.05, df=10, p=0.96).
Discussion
We found low receptor-bound [3H]pirenzepine levels in prefrontal cortex from patients who had schizophrenia. This low [3H]pirenzepine binding is consistent with the low binding of [3H]pirenzepine in the caudate-putamen (39) and hippocampal formation (25) that was found in earlier studies.
Antipsychotic drug treatment is nearly ubiquitous in patients with schizophrenia, and antiparkinsonian drugs such as benztropine are commonly administered for the control of extrapyramidal side effects. Subsequently, neuroleptic agents are potential confounders in many neurobiological studies of schizophrenia (40).
Although we are unable to unequivocally exclude a causal relationship between antipsychotic drug treatment and low receptor-bound [3H]pirenzepine levels in the prefrontal cortex of schizophrenia patients, preliminary studies suggest that treatment with antipsychotic drugs did not confound the measurement of receptors in schizophrenia. In our rat studies, treatment with typical or atypical antipsychotic drugs tended to increase or have no effect on the levels of receptor binding. As the drugs presently considered are putative muscarinic antagonists (41, 42), they would not be expected to directly down-regulate muscarinic receptor levels. However, the possibility that receptor levels are reduced through more complex mechanisms in human brains than in rat brains cannot be excluded.
In regard to the benztropine studies, while the rat data indicate no effect of drug administration on receptor-bound [3H]pirenzepine levels, a consideration of patient drug treatment revealed a more complex picture. The difference in radioligand binding from comparison values was greater for the schizophrenia patients who had been treated with benztropine than for those who had not received benztropine. However, the radioligand binding of the non-benztropine-treated patients was significantly lower in Brodmann’s areas 9 and 46, and nonsignificantly lower in Brodmann’s areas 8 and 10, than in the comparison subjects. We previously reported (39) that the lower-than-normal [3H]pirenzepine binding in caudate-putamen from schizophrenia patients was greater for patients treated with benztropine than for non-benztropine-treated patients. Moreover, as in the present findings, the density of [3H]pirenzepine binding to caudate-putamen for the non-benztropine-treated schizophrenia group was also lower than that for the comparison group. With the limitations of our drug studies, there are a number of possible explanations for the present findings (39). One possibility is that although treatment of patients with benztropine may cause a down-regulation of muscarinic receptor binding, benztropine is not the sole cause of the reduced receptor binding. It is important to note that in the event that altered radioligand binding reflects altered receptor levels, a muscarinic receptor antagonist such as benztropine would not be expected to directly down-regulate muscarinic receptor levels. However, a drug-induced decrease in receptor levels through other mechanisms cannot be excluded. A second possibility is supported by the rat studies, which indicate that benztropine treatment does not induce a decrease in muscarinic receptor binding. Perhaps the observed relationship between lower radioligand binding levels in the human prefrontal cortex and treatment with benztropine reflects a propensity to develop extrapyramidal side effects and a concomitant need for treatment after alteration of muscarinic receptor activity (39).
While we can only speculate on the import of the present findings, it seems reasonable to suggest that the muscarinic system may be involved in the pathology and/or pharmacotherapeutics of schizophrenia. First, in the event that altered muscarinic receptor binding in schizophrenia is influenced by antipsychotic or antiparkinsonian drug treatment, this may be relevant to understanding the mechanisms of current and future pharmacotherapeutic strategies. Second, a possible primary change of cortical muscarinic receptors in the pathology of schizophrenia may be subsequent to a pre- or posttranscriptional abnormality. Studies of M1 and M4 receptor transcripts may be useful in determining aberrant receptor gene expression. Finally, decreased radioligand binding may reflect a down-regulation in M1 and/or M4 receptor levels after an increase in cholinergic efflux following overactivity of the cholinergic basal forebrain system. If this is the case, on the basis of the putative dopaminergic modulation of transmission in the basal forebrain mediated by γ-aminobutyric acid (GABA), a hypercholinergic model may be relevant to the dopamine hypothesis of schizophrenia (43–45). It is interesting that dopamine receptor stimulation in the nucleus accumbens has been shown to reduce the activity of inhibitory GABA-ergic efferents to the basal forebrain and increase the excitability of cholinergic basal-forebrain corticopetal neurons (46–48). Thus, increased mesolimbic dopaminergic transmission, deemed central to schizophrenia (49), may induce increased cortical cholinergic efflux and down-regulate postsynaptic M1 and M4 receptor levels and the binding presently measured. This model is consistent with the current understanding of the actions of antipsychotic drugs such as haloperidol. Haloperidol normalizes dopamine-mediated transmission in the nucleus accumbens, possibly through dopamine D2 receptors, and hence reduces the excitability of cortical cholinergic input (48, 50). Accordingly, the present rat studies suggest that long-term treatment with haloperidol induces up-regulation of levels of [3H]pirenzepine-labeled receptors in the rat frontal cortex.
In conclusion, these studies indicate an abnormality of the muscarinic receptor system in the prefrontal cortex in schizophrenia. Our earlier findings of low muscarinic receptor binding in the caudate-putamen (39, 51) and hippocampal formation (25) in schizophrenia suggest that the abnormalities presently measured might not be limited to the prefrontal cortex. Although the interpretation of the data presented here remains tentative, these findings may be important in elucidating the cascade of events involved in the abnormal neurobiology of schizophrenia, and they may provide insight into current pharmacological interventions that could prove useful to the development of new strategies for the treatment of schizophrenia.
 |
 |
 |
Received Aug. 14, 2000; revision received Nov. 20, 2000; accepted Dec. 20, 2000. From the Division of Molecular Schizophrenia, Rebecca L. Cooper Research Laboratories, the Mental Health Research Institute of Victoria, Parkville, Victoria, Australia; and the Department of Pathology, University of Melbourne, Parkville, Victoria, Australia. Address reprint requests to Dr. Crook, Department of Otolaryngology, Royal Victorian Eye and Ear Hospital, 32 Gisborne St., East Melbourne, Victoria 3002, Australia. Supported in part by a National Health and Medical Research Council Dora Lush Post-Graduate Scholarship to Dr. Crook, by a Young Investigator award from the National Alliance for Research on Schizophrenia and Depression to Dr. Dean, and by the Rebecca L. Cooper Research Foundation.

Figure 1. Representative Images of the Binding of [3H]Pirenzepine in Brodmann’s Areas 8, 9, 10, and 46 of the Human Prefrontal Cortex and in Rat Frontal Cortex

Figure 2. Binding of [3H]Pirenzepine in Brodmann’s Areas 8, 9, 10, and 46 of Prefrontal Cortex From Deceased Patients With Schizophrenia Who Had or Had Not Received Benztropine and From Healthy Comparison Subjects
aSignificant difference among groups according to ANCOVA controlling for freezer time (F=8.55, df=2, 33, p<0.001), nonsignificant difference between non-benztropine-treated patients and comparison subjects (p=0.29, least significant difference test), and significant difference between the two patient groups (p=0.005, least significance difference test).
bSignificant difference among groups according to ANCOVA controlling for freezer time (F=12.29, df=2, 33, p<0.0001) and significant differences between the non-benztropine-treated patients and both the comparison subjects (p=0.04) and the benztropine-treated patients (p=0.004) (least significance difference tests).
cSignificant difference among groups according to ANCOVA controlling for freezer time (F=10.81, df=2, 33, p<0.0002), nonsignificant difference between non-benztropine-treated patients and comparison subjects (p=0.10, least significant difference test), and significant difference between the two patient groups (p=0.004, least significant difference test).
dSignificant difference among groups according to ANCOVA controlling for freezer time (F=23.67, df=2, 33; p<0.001) and significant differences between the non-benztropine-treated patients and both the comparison subjects (p=0.007) and the benztropine-treated patients (p=0.0001) (least significant difference tests).
1. Zilles K: Cortex, in The Human Nervous System. Edited by Paxinos G. San Diego, Academic Press, 1990, pp 757–802Google Scholar
2. Heckers S: Neuropathology of schizophrenia: cortex, thalamus, basal ganglia, and neurotransmitter-specific projection systems. Schizophr Bull 1997; 23:403–421Crossref, Medline, Google Scholar
3. Glantz LA, Lewis DA: Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000; 57:65–73Crossref, Medline, Google Scholar
4. Glantz LA, Lewis DA: Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia: regional and diagnostic specificity. Arch Gen Psychiatry 1997; 54:943–952Crossref, Medline, Google Scholar
5. Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WS: Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: a possible neurochemical basis for “hypofrontality.” Mol Psychiatry 1999; 4:39–45Crossref, Medline, Google Scholar
6. Honer WG, Falkai P, Chen C, Arango V, Mann JJ, Dwork AJ: Synaptic and plasticity-associated proteins in anterior frontal cortex in severe mental illness. Neuroscience 1999; 91:1247–1255Google Scholar
7. Goldman-Rakic PS: Psychopathology and neuropathology of prefrontal cortex in schizophrenia, in Schizophrenia. Edited by Fog R, Gerlach J, Hemmingsen R. Copenhagen, Munksgaard, 1995, pp 126–136Google Scholar
8. Park S, Holzman PS: Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry 1992; 49:975–982Crossref, Medline, Google Scholar
9. Mesulam M, Hersh LB, Mash DC, Geula C: Differential cholinergic innervation within functional subdivisions of the human cerebral cortex: a choline acetyltransferase study. J Comp Neurol 1992; 318:316–328Crossref, Medline, Google Scholar
10. Bigl V, Woolf NJ, Butcher LL: Cholinergic projections from the basal forebrain to frontal, parietal, temporal, occipital, and cingulated cortices: a combined fluorescent tracer and acetylcholinesterase analysis. Brain Res Bull 1982; 8:727–749Crossref, Medline, Google Scholar
11. Saper CB: Organization of cerebral cortical afferent systems in the rat, II, magnocellular basal nucleus. J Comp Neurol 1984; 222:313–342Crossref, Medline, Google Scholar
12. Sarter M, Bruno JP: Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res 1997; 23:28–46Crossref, Google Scholar
13. Mesulam MM: Behavioral neuroanatomy of cholinergic innervation in the primate cerebral cortex. EXS 1989; 57:1–11Medline, Google Scholar
14. Buzsaki G, Gage FH: The cholinergic nucleus basalis: a key structure in neocortical arousal. EXS 1989; 57:159–171Medline, Google Scholar
15. Everitt BJ, Robbins TW: Central cholinergic systems and cognition. Annu Rev Psychol 1997; 48:649–684Crossref, Medline, Google Scholar
16. Smith G: Animal models of Alzheimer’s disease: experimental cholinergic denervation. Brain Res 1988; 472:103–118Crossref, Medline, Google Scholar
17. Kato H, Aikawa H, Shinohara Y: The effects of lesions in the nucleus basalis of Meynert and physostigmine on rat frontal cortex acetylcholine level. Tokai J Exp Med 1999; 24:125–130Medline, Google Scholar
18. Rossner S, Schliebs R, Perez-Polo JR, Wiley RG, Bigl V: Differential changes in cholinergic markers from selected brain regions after specific immunolesion of the rat cholinergic basal forebrain system. J Neurosci Res 1995; 40:31–43Crossref, Medline, Google Scholar
19. Holmes TC, Nitsch RM, Erfurth A, Wurtman RJ: Phospholipid and phospholipid metabolites in rat frontal cortex are decreased following nucleus basalis lesions. Ann NY Acad Sci 1993; 695:241–243Crossref, Medline, Google Scholar
20. Katayama S, Kito S, Yamamura Y: Increase of muscarinic receptor following kainic acid lesions of the nucleus basalis magnocellularis in rat brain: an autoradiographic study. Res Commun Chem Pathol Pharmacol 1990; 68:391–394Medline, Google Scholar
21. Fuji K, Hiramatsu M, Hayashi S, Kameyama T, Nabeshima T: Effects of propentofylline, a NGF synthesis stimulator, on alterations in muscarinic cholinergic receptors induced by basal forebrain lesion in rats. Neurosci Lett 1993; 150:99–102Crossref, Medline, Google Scholar
22. Haroutunian V, Mantin R, Kanof PD: Frontal cortex as the site of acton of physostigmine in nbM-lesioned rats. Physiol Behav 1990; 47:203–206Crossref, Medline, Google Scholar
23. Haroutunian V, Kanof PD, Tsuboyama G, Davis KL: Restoration of cholinomimetic activity by clonidine in cholinergic plus noradrenergic lesioned rats. Brain Res 1990; 507:261–266Crossref, Medline, Google Scholar
24. Levey AI: Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA 1996; 93:13541–13546Google Scholar
25. Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B: Decreased muscarinic receptor binding in subjects with schizophrenia: a study of the human hippocampal formation. Biol Psychiatry 2000; 48:381–388Crossref, Medline, Google Scholar
26. Neeper R, Richelson E, Nelson A: Neuroleptic binding to muscarinic M2 receptors of normal human heart in vitro and comparison with binding to M1 and dopamine D2 receptors of brain. Neuropharmacology 1991; 30:527–529Crossref, Medline, Google Scholar
27. Bolden C, Cusack B, Richelson E: Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells. J Pharmacol Exp Ther 1992; 260:576–580Medline, Google Scholar
28. Bolden C, Cusack B, Richelson E: Clozapine is a potent and selective muscarinic antagonist at the five cloned human muscarinic acetylcholine receptors expressed in CHO-K1 cells. Eur J Pharmacol 1991; 192:205–206Crossref, Medline, Google Scholar
29. Bymaster FP, Nelson DL, DeLapp NW, Falcone JF, Eckols K, Truex LL, Foreman MM, Lucaites VL, Calligaro DO: Antagonism by olanzapine of dopamine D1, serotonin2, muscarinic, histamine H1 and α1-adrenergic receptors in vitro. Schizophr Res 1999; 37:107–122Crossref, Medline, Google Scholar
30. Antiparkinsonain agents, in MIMS Annual. Edited by Wilkinson LL. Sydney, IMS Publishing, 1989, pp 4–193Google Scholar
31. Brown JH: Atropine, scopolamine, and related antimuscarinic drugs, in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 8th ed. Edited by Gilman AG, Rall TW, Nies AS, Taylor P. New York, Pergamon Press, 1990, pp 150–165Google Scholar
32. Hulme EC, Birdsall NJM, Buckley NJ: Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol 1990; 30:633–673Crossref, Medline, Google Scholar
33. Hill C, Keks N, Roberts S, Opeskin K, Dean B, MacKinnon A, Copolov D: Problem of diagnosis in postmortem brain studies of schizophrenia. Am J Psychiatry 1996; 153:533–537Link, Google Scholar
34. Foster P: Neuroleptic equivalence. Pharmaceutical J 1998; 243:431–432Google Scholar
35. Kingsbury AE, Foster OJ, Nisbet AP, Cairns N, Bray L, Eve DJ, Lees AJ, Marsden CD: Tissue pH as an indicator of mRNA preservation in human postmortem brain. Mol Brain Res 1995; 28:311–318Crossref, Medline, Google Scholar
36. Brodmann K: Description of individual brain maps, in Brodmann’s “Localisation in the Cerebral Cortex.” Edited by Garey LJ. London, Smith-Gordon, 1994, pp 107–174Google Scholar
37. Acri JB, Siedleck BK, Witkin JM: Effects of benztropine on behavioral and toxic effects of cocaine: comparison with atropine and the selective dopamine uptake inhibitor 1-[2-(diphenylmethoxy)ethyl]-4-(3-phenyl-propyl)-piperazine. J Pharmacol Exp Ther 1996; 277:198–206Medline, Google Scholar
38. Paxinos G, Watson C: The Rat Brain in Stereotaxic Coordinates, 2nd ed. Sydney, New South Wales, Australia, Academic Press, 1986Google Scholar
39. Dean B, Crook JM, Opeskin K, Hill C, Keks N, Copolov DL: The density of muscarinic M1 receptors is decreased in the caudate-putamen of subjects with schizophrenia. Mol Psychiatry 1996; 1:54–58Medline, Google Scholar
40. Harrison P: The neuropathological effects of antipsychotic drugs. Schizophr Res 1999; 40:87–99Crossref, Medline, Google Scholar
41. Richelson E: Preclinical pharmacology of neuroleptics: focus on new generation compounds. J Clin Psychiatry 1996; 57(suppl 11):4–11Google Scholar
42. Snyder SH, Greenberg D, Yamumura HI: Antischizophrenic drugs: affinity for muscarinic cholinergic receptor sites in the brain predicts extrapyramidal effects. J Psychiatr Res 1974; 11:91–95Crossref, Medline, Google Scholar
43. Snyder SH: Catecholamines in the brain as mediators of amphetamine psychosis. Arch Gen Psychiatry 1972; 27:169–179Crossref, Medline, Google Scholar
44. Gray JA, Joseph MH, Hemsley DR, Young AM, Warburton EC, Boulenguez P, Grigoryan GA, Peters SL, Rawlins JN, Taib CT, Yee BK, Cassaday H, Gal G, Gusak O, Joel D, Shadach E, Shalev U, Tarrasch R, Feldon J: The role of mesolimbic dopaminergic and retrohippocampal afferents to the nucleus accumbens in latent inhibition: implications for schizophrenia. Behav Brain Res 1995; 71:19–31Crossref, Medline, Google Scholar
45. Yang CR, Seamans JK, Gorelova N: Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology 1999; 21:161–194Crossref, Medline, Google Scholar
46. Bourdelais A, Kalivas P: Apomorphine decreases extracellular GABA in the ventral pallidum of rats with 6-OHDA lesions in the nucleus accumbens. Brain Res 1992; 577:306–311Crossref, Medline, Google Scholar
47. Moore H, Fadel J, Sarter M, Bruno JP: Role of accumbens and cortical dopamine receptors in the regulation of cortical acetylcholine. Neuroscience 1999; 88:811–822Crossref, Medline, Google Scholar
48. Moore H, Stuckman S, Sarter M, Bruno JP: Stimulation of cortical acetylcholine efflux by FG 7142 measured with repeated microdialysis sampling. Synapse 1995; 21:324–331Crossref, Medline, Google Scholar
49. Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D: Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 1997; 94:2569–2574Google Scholar
50. Sarter M, Bruno JP: Abnormal regulation of corticopetal cholinergic neurons and impaired information processing in neuropsychiatric disorders. Trends Neurosci 1999; 22:67–74Crossref, Medline, Google Scholar
51. Crook JM, Dean B, Pavey G, Copolov DL: The binding of [3H]AF-DX 384 is reduced in the caudate-putamen of subjects with schizophrenia. Life Sci 1999; 64:1761–1771Google Scholar

