
Increased Glutamatergic Neurotransmission and Oxidative Stress After Alcohol Withdrawal
Abstract
OBJECTIVE: Neurophysiological and pathological effects of ethanol may be mediated, to an important extent, via the glutamatergic system. Animal studies indicate the acute effects of ethanol disrupt glutamatergic neurotransmission by inhibiting the response of the N-methyl-D-aspartate (NMDA) receptor. Persistent attenuation of glutamatergic neurotransmission by chronic ethanol exposure results in the compensatory up-regulation of NMDA receptors. Whether glutamatergic neurotransmission and oxidative stress are enhanced during ethanol withdrawal in humans is unknown. METHOD: CSF was obtained from 18 matched comparison subjects and from 18 patients with alcohol dependence 1 week and 1 month after cessation of ethanol ingestion. CSF samples were analyzed for excitatory neurotransmitters, γ-aminobutyric acid (GABA), and markers for oxidative stress. RESULTS: The alcohol-dependent patients' CSF levels of aspartate, glycine, and N-acetylaspartylglutamate were all higher than those of the comparison subjects, and their concentration of GABA was lower. In addition, there were significant correlations between excitatory neurotransmitters and oxidative stress markers, which suggest that the two mechanisms may play an interactive role in neurotoxicity mediated by ethanol withdrawal. CONCLUSIONS: The data suggest that augmentation of excitatory neurotransmission may lead to enhanced oxidative stress, which, in concert with reduced inhibitory neurotransmission, may contribute to the symptoms of ethanol withdrawal and associated neurotoxicity in humans. Whether these abnormalities represent a trait- or state-dependent marker of ethanol dependence remains to be resolved.
Ethanol has long been considered as a pharmaco~logical depressant of the central nervous system (1). It was suggested 20 years ago that alcohol may suppress the excitability of neurons by inhibiting the receptors for the excitatory transmitter glutamate (2). More than a decade later, Lovinger et al. (3) demonstrated that ethanol in clinically relevant concentrations (between 5 and 50 mM) produces a dose-dependent inhibition of currents induced by the glutamate agonist N-methyl-D-aspartate (NMDA) in hippocampal neurons. Other studies (4–7) also showed that ethanol acutely disrupts glutamatergic neurotransmission by inhibiting the response of NMDA receptors. Furthermore, accumulating evidence from animal studies suggests that the neurophysiological and pathological effects of ethanol are mediated to a considerable extent via the glutamatergic system (see review by Tsai et al. [8]). This attenuation of excitatory neurotransmission by ethanol may be exacerbated by its known enhancement of inhibitory γ-aminobutyric acid (GABA) neurotransmission (9).
One of the adaptive consequences of chronic inhibition of glutamatergic neurotransmission by prolonged ethanol exposure is the compensatory up-regulation of NMDA receptors (10–13). Thus, upon the removal of ethanol's inhibitory effect, as would occur during alcohol withdrawal and early abstinence, one would expect to see increased excitatory neurotransmission. Several lines of evidence support this hypothesis. NMDA exacerbates ethanol withdrawal seizures in experimental animals, while MK-801 and other NMDA receptor antagonists decrease the occurrence and severity of ethanol withdrawal seizures (2, 12, 14, 15). Brain structures activated during alcohol withdrawal exhibit increased expression of the immediate-early gene c-fos mRNA, which is abolished by MK-801 (16). More recently, increased extracellular concentrations of excitatory ami~no acids have been demonstrated in rat hippocampus during ethanol withdrawal (17).
To our knowledge, studies with human subjects on the role of excitatory neurotransmission after ethanol withdrawal have not been conducted. Increased excitatory neurotransmission in humans may, however, be inferred from acamprosate's anticraving effect for alcohol (18). This structural analog of glutamate has been demonstrated to reduce the activation of NMDA receptor ion channels. Thus, acamprosate is thought to exert its therapeutic effects by attenuating the neuronal hyperexcitability induced by chronic ethanol consumption and withdrawal (19).
The present study was designed to quantify indices of both excitatory and inhibitory amino acid neurotransmission in patients with alcohol dependence at 1 week and 1 month after alcohol withdrawal. Markers of oxidative stress were also measured to elucidate their pathophysiological importance since excessive activation of glutamate receptors causes oxidative stress (20). We hypothesized that excitatory neurotransmission is enhanced after ethanol withdrawal in humans.
METHOD
CSF was obtained from 18 patients who fulfilled the DSM-III-R diagnosis of alcohol dependence; they comprised four women and 14 men and had a mean age of 37.5 years (SD=8.4, range=27–53). Their drinking was in the moderately severe range; they had a score on the CAGE (21) of 3.1 (SD=1.0) and a score on the Michigan Alcoholism Screening Test (22) of 40.9 (SD=9.7). They had been drinking for an average of 20.7 years (SD=9.1); the total amount of absolute ethanol consumed in the last 6 months was 24.7 kg (SD=17.4), and the lifetime amount was 417.7 kg (SD=259.3). None of the patients had a history of withdrawal or other seizures. Other than ethanol and nicotine dependence (11 subjects), the study group did not have any history of substance abuse in the 6 months before admission to the National Institute on Alcohol Abuse and Alcoholism (NIAAA). None of the patients had any lifetime use of intravenous drugs or crack cocaine.
There was no evidence of cirrhosis or decompensated liver disease in the subjects. Mildly elevated levels of liver function indices were shown in laboratory tests performed upon admission to the study. The mean for alanine aminotransferase was 50.4 U/liter (normal range=6–41), the mean for aspartate aminotransferase was 57.3 U/liter (normal=9–34), and the mean for γ-glutamyltransferase was 114.6 U/liter (normal=14–84); all were consistent with recent drinking. Despite the level and duration of drinking in this group, there was no overt laboratory or clinical evidence of hypomagnesemia, myopathy, anemia, severe malnutrition, or infection, indices of common complications of chronic alcoholism.
Two CSF samples were acquired by lumbar puncture at 8.6 days (SD=1.9) and 33.0 days (SD=2.2) of abstinence from ethanol. Patients were admitted to the NIAAA research ward and enrolled in the study 1 week before the first lumbar puncture. During the period of study, the patients did not receive any psychotropic medication, including benzodiazepines. For the first 24 hours after admission, scores on the Clinical Institute Withdrawal Assessment Alcohol Scale (23) were monitored hourly. Any patients with sustained scores of 12 or above were treated with benzodiazepines and excluded from the study. Although this procedure excluded patients with severe alcohol withdrawal, we wished to avoid the introduction of a factor (benzodiazepine medication) with a high likelihood of influencing the CSF measures. We did not attempt to perform lumbar puncture during the acute withdrawal phase. Rather, our study was designed to investigate subacute alcohol withdrawal and the intermediate postwithdrawal period.
Comparison CSF was obtained from age-matched subjects, three women and 15 men with a mean age of 38.3 years (SD=9.0, range=27–54). Subjects who had a history of neurological disorders, psychiatric disorders, or major medical illness were excluded. After complete description of the study to the subjects, written informed consent was obtained. The study was approved by the NIAAA intramural program institutional review board.
Both the alcoholic subjects and healthy comparison subjects were placed on a low-monoamine diet at least 3 days before the lumbar puncture. All subjects fasted after midnight and were kept at strict bed rest the morning of lumbar puncture. Lumbar puncture was performed with the subjects in the left lateral decubitus position between 9:00 and 10:00 a.m. CSF was collected by 1-ml fractions. The same fractions of the CSF were subjected to the assays described in the following sections.
Amino Acids and Peptides
Amino acids were measured by o-phthalaldehyde precolumn derivatization coupled with a reverse-phase C-18 column for high-performance liquid chromatography separation and fluorescent detection (24). The absolute concentrations of the amino acids were determined by computer analysis of peak height with internal and external standards. N-Acetylaspartylglutamate (NAAG) and N-acetylaspartate (NAA) were measured by anion-exchange high-performance liquid chromatography separation and ultraviolet detection at a wavelength of 214 nm (25). Their concentrations were determined by peak height calculation as well.
Superoxide Dismutase
Total superoxide dismutase activity in CSF was analyzed by a modification of a spectrophotometric method (26). The reduction of cytochrome c was used to detect and measure superoxide dismutase activity. The reduction of acetylated cytochrome c by ·O2– is accompanied by a change in absorbance at 550 nm. Aliquots of CSF were added to 20 mM bicarbonate buffer, containing 10 µM sodium azide, 10 µM cytochrome c, 100 µM xanthine, and 1 mM EDTA. The reduction of acetylated cytochrome c was initiated by the addition of xanthine oxidase.
Lipid Hydroperoxides
The levels of lipid hydroperoxides were measured according to an adaptation of the ferrous oxidation/xylenol orange method described by Puttfarcken et al. (27) by using 10 µl of CSF.
Protein Carbonyl
The method for protein carbonyl assay was adapted from the one described by Vevine et al. (28), and we used 90 µl of CSF.
Statistics
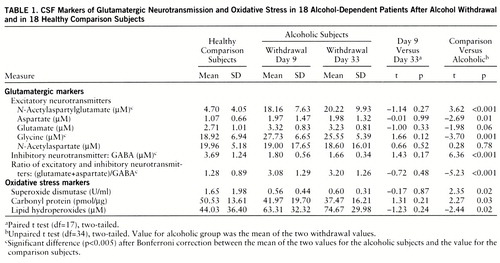
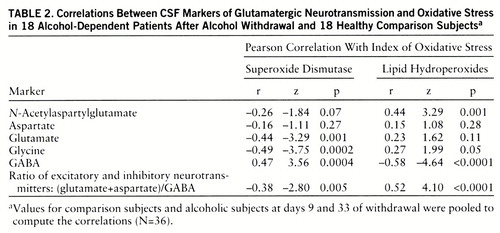
We first performed paired t tests (two-tailed) of the differences between days 9 and 33 and discovered no significant change in the neuro~chemical measures between these two time points (table 1). We next derived means for days 9 and 33. The means for these two time points were compared to the values of the normal comparison subjects by unpaired t tests (two-tailed). Because of the multiple analyses, Bonferroni correction was applied (table 1). To estimate their contributions to the variance, we computed Pearson coefficients for the correlations between the markers of oxidative stress and the glutamatergic markers.
RESULTS
Table 1 lists the mean NAAG, amino acid (NAA, aspartate, glutamate, glycine, GABA), protein carbonyl group, and lipid hydroperoxide levels and the superoxide dismutase activity in CSF of the healthy comparison subjects and the patients after alcohol withdrawal. The CSF concentrations of glycine and NAAG of the alcohol-dependent patients were significantly higher than the levels of the healthy comparison subjects at both points after ethanol withdrawal (glycine: day 9, 47%; day 33, 35%; NAAG: day 9, 286%; day 33, 330%). The differences in CSF aspartate and glutamate levels after alcohol with~drawal were not significantly different from the comparison subjects' levels after Bonferroni correction (aspartate: day 9, 84%; day 33, 85%; glutamate: day 9, 23%; day 33, 19%). In contrast, the GABA levels after alcohol withdrawal were significantly lower than the comparison values (day 9, –51%; day 33, –55%). However, the NAA concentrations did not differ between the healthy comparison subjects and the alcoholics. Notably, these effects persisted until the second measurement at about 1 month after the withdrawal from alcohol.
After Bonferroni correction, the higher concentration of lipid hydroperoxides (day 9, 44%; day 33, 70%) in the CSF of the patients who had experienced alcohol withdrawal and their lower superoxide dismutase activity (day 9, –66%; day 33, –64%) were not significantly different from the values for the comparison subjects (table 1). These changes were of the same magnitude in the second measure. The protein carbonyl content did not different significantly between the comparison and alcoholic subjects.
To investigate the relationship between excitatory neurotransmission and oxidative stress, we pooled the data to compute the correlations between the oxidative stress markers—superoxide dismutase activity and lipid hydro~peroxide level—and the excitatory neurotransmitters (table 2). The correlation coefficients were remarkable for the negative correlations of the excitatory neurotransmitter markers with superoxide dismutase and the positive correlations with lipid hydro~peroxide levels. In contrast, the concentration of the inhibitory neurotransmitter, GABA, was positively correlated with superoxide dismutase but negatively correlated with lipid hydroperoxides (table 2). Similar but more robust correlations between the oxidative stress indices and excitatory neurotransmission were observed when the status of excitatory neurotransmission was expressed as the ratio of excitatory to inhibitory amino acid neurotransmitters: (glutamate+aspartate)/GABA.
DISCUSSION
The chronically alcohol-dependent patients in this study who underwent mild to moderate alcohol withdrawal (scores less than 12 on the Clinical Institute Withdrawal Assessment Alcohol Scale) had higher concentrations of CSF indices of excitatory neurotransmission, NAAG and glycine, than did healthy comparison subjects (table 1, figure 1). We considered glycine to be a marker of excitatory neurotransmission because of its role as a coagonist of the NMDA receptor (29), although it can also serve as an inhibitory neurotransmitter in the spinal cord (30).
Since we do not have data from CSF obtained before alcohol withdrawal, we can not ascertain whether the differences that we observed were state dependent (due to withdrawal) or trait related (due to alcoholism). The values of glutamate, glycine, and NAAG observed in this cohort of comparison subjects were similar to those in comparison CSF from another study in this laboratory (31), suggesting that the differences do not reflect a bias in the comparison samples. The patients' measurements during alcohol intoxication and after the patients presumably returned to their baselines after prolonged abstinence are not available. These present findings may either have preceded the alcoholism itself or be the consequence of long-term alcoholism, not directly associated with the state of alcohol withdrawal.
The relative contributions of inhibitory and excitatory pools of glycine in CSF are not clear. It is also unclear how the low-monoamine diet may have affected the neurochemical markers studied. However, it is conceivable that the stable environment and regular diet can decrease oxidative stress, and hence attenuate excitatory neurotransmission (32), in alcoholics, who often encounter dietary deficiencies and various medical complications. Therefore, our findings of greater than normal excitatory neurotransmission after alcohol withdrawal are more likely due to either the withdrawal state per se or the preexisting condition of chronic alcoholism, which persisted into the withdrawal state, than to the diet itself.
Symptoms of alcohol withdrawal can last long after cessation of alcohol use. In our patients the neurochemical changes persisted up to 1 month of abstinence (table 1). To the extent that the concentrations in the CSF represent synaptic events, the neurochemical differences may reflect intermediate- and long-term effects of alcohol withdrawal after moderately severe alcohol consumption. However, it will be important to study the neurochemical changes during acute withdrawal, which may reveal larger changes, since the NMDA neuro~transmission up-regulation is reported to be most robust during the first 48 hours of withdrawal in animals (12). In vivo magnetic resonance spectroscopy can also help to determine the extent of enhanced excitatory neurotransmission during acute withdrawal. Alternatively, it is plausible that patients who experience symptoms of alcohol withdrawal are vulnerable to withdrawal because of their enhanced excitatory neurotransmission at baseline. Further studies of at-risk and long-term abstinent populations need to address this question directly. If indeed the enhanced excitatory neurotransmission is a trait marker, it may help to identify a group at high risk for alcoholism. In either case, our findings support the proposal that the pathophysiological basis for the symptoms of alcohol withdrawal and alcohol-induced neurotoxicity may be mediated by enhanced excitatory neurotransmission.
In addition to high indices of excitatory neurotransmission, we also observed a low CSF GABA concentration during alcohol withdrawal. It is well established that GABAergic neurotransmission is also an important component in the neurobiological effect of alcohol because of its activation of GABA-gated chloride channels (9). The combined effect of the inferred increased excitatory neurotransmission and decreased GABAergic neurotransmission would lead to a substantial amplification of the overall excitatory neurotransmission, as indicated by the ratio (aspartate+glutamate)/GABA (table 1). The only previous study of CSF GABA concentrations among alcoholic patients from this research program did not demonstrate a change of GABA concentration after cessation of alcohol use (33). This apparent discrepancy may be due to the different duration of abstinence in the subjects, which was longer in the previous study. In animal studies on the effect of extended exposure to alcohol on GABAergic function, a reduced coupling between the GABA recognition site on the GABAA receptor and the chloride channel has been found (34). Thus, alcohol withdrawal may be associated with a more serious impairment of inhibitory neurotransmission than indicated by CSF GABA concentrations. The coexistence of low inhibitory and high excitatory neurotransmission revealed in our CSF study needs to be investigated in neuroanatomical studies that address the neuronal circuitry of the glutamate and GABA systems. For example, the inhibitory feedback by cortical GABAergic interneurons to the glutamatergic neurons is one of the candidates to investigate.
Even though the patients experienced only mild to moderate alcohol withdrawal symptoms, the group that we studied had a long history of alcohol dependence. Chronic attenuation of glutamatergic neurotransmission by ethanol results in a compensatory up-regulation of NMDA receptors that persists after withdrawal (10–13, 35). Augmentation of glycine, a coagonist of NMDA receptors, can further enhance glutamatergic neurotransmission (29). Glycine also acts as an inhibitory neurotransmitter at the strychnine-sensitive glycine receptor, which is concentrated primarily in the spinal cord (30). Glycine sites on the NMDA receptor may not be saturated (36). Thus, a higher glycine concentration may augment NMDA receptor function in cortical structures. Overall, the combined augmentation of the synaptic concentration of excitatory neurotransmitters during alcohol withdrawal, in concert with the up-regulation of the postsynaptic receptors, may contribute to the enhanced glutamatergic neurotransmission. Consistent with this inference, glutamate release is significantly enhanced in alcohol-preferring rats (37). During ethanol withdrawal, dependent mice have 50%–70% more brain MK-801 binding sites, a marker for NMDA receptors, than nondependent control mice (10). Ethanol-naive mice prone to withdrawal seizures have also been found to have more MK-801 binding sites in the hippocampus (38). In addition, NMDA exacerbates, while MK-801 and other NMDA receptor antagonists attenuate, the occurrence and severity of seizures associated with withdrawal of ethanol after chronic intake (2, 15).
We also found high CSF NAAG levels after ethanol withdrawal (table 1). Physiological studies (39–43) have demonstrated that NAAG blocks the action of endogenous glutamate. However, NAAG is catabolized to glutamate and NAA by the specific high-affinity membrane-bound surface peptidase N-acetylated-α-linked acidic dipeptidase (NAALADase) (44). Through the action of NAALADase, NAAG may serve as a precursor to extrasynaptic glutamate. Furthermore, NAAG is an agonist at the metabotropic receptor mGLUR-3, which inhibits glutamate release (45). In addition to the glutamatergic neurons, NAAG is also colocalized in subpopulations of monoaminergic and cholinergic neurons (46). Thus, aminergic activation during alcohol withdrawal may contribute to the increased release of NAAG in the CSF. Whether the elevated levels of NAAG represent a compensatory protective mechanism against glutamatergic damage through its inhibition of NMDA receptors and glutamate release or a source of increased extrasynaptic glutamate remains unresolved.
We observed higher than normal lipid peroxide levels and low superoxide dismutase activity in the CSF of alcohol-withdrawn patients (table 1). The differences, however, did not reach statistical significance after the Bonferroni correction. Bonferroni correction is applied to variables independent of each other. The neurochemical variables we measured are not independent of each other. Not only are oxidative stress markers related to each other (low superoxide dismutase activity can lead to high levels of lipid hydroperoxide and protein carbonyl), but glutamatergic markers are also related to each other. NAAG is the precursor of NAA, glutamate, and aspartate (47). Glutamate and aspartate are regulated together in most excitotoxic conditions (31). GABA is a product of glutamate through the action of glutamate decarboxylase. More than that, oxidative stress and glutamatergic neurotransmission are mutually enhancing (20, 32). To balance between type 1 and type 2 errors, the physiological meaning of the changes in superoxide dismutase activity and lipid peroxide level need to be discussed.
There are many potential mechanisms by which ethanol can generate free radicals, leading to lipid peroxidation. Ethanol itself can induce a dose-dependent increase in lipid peroxidation in brain homogenates. Lipid peroxidation is related to the metabolism of ethanol and acetaldehyde by secondary pathways that are known to generate oxygen-related free radicals (48). Also, formation of long-lived hydroxyethyl free radicals from ethanol may result in membrane damage due to an increase in lipid peroxidation (49). In addition to lipid peroxidation, ethanol-fed rats display increases in the production of thiobarbituric acid-reactive material, the generation of H2O2, and OH·-like species (50, 51). Formation of free radicals from ethanol is also mediated by a transition-metal- and cytochrome-P450-derived superoxide-dependent oxidizing species (52). Ethanol-induced cytochrome P450 IIE1 is constitutively expressed in the central nervous system. The induction of P450 IIE1 may lead to increased generation of reactive oxygen species and hydroxyethyl radicals, contributing to oxidative stress in the brains of chronic alcoholics (53, 54). Nevertheless, the mechanism and physiological implications of the trend toward lower protein carbonyl during alcohol withdrawal (table 1) remain to be explored.
Superoxide dismutase is an enzyme critical in the detoxification of superoxide (55). Generation of free radicals by ethanol can be prevented by superoxide dismutase (56), suggesting that superoxide is the primary source for other oxyradicals resulting from ethanol administration. Our study revealed a long-lasting reduction in superoxide dismutase activity in CSF after alcohol withdrawal. When superoxide dismutase activity is decreased, neurons are more vulnerable to oxyradical injury, and this is consistent with the high levels of lipid hydroperoxides that we observed (r=–0.30, p=0.03). Parallel with our findings, reduced activity of superoxide dismutase has been found in pigs chronically fed ethanol (57). In addition to the attenuation of superoxide dismutase activity, the antioxidants glutathione and vitamin E were also shown to be decreased by ethanol exposure, which can further weaken the defense against oxidative stress (58, 59).
The negative correlations between CSF levels of excitatory neurotransmitters and superoxide dismutase activity and the positive correlations with lipid hydroperoxide levels suggest an important relationship between enhanced excitatory neurotransmission and oxidative damage after ethanol withdrawal (figure 1). The activation of glutamate ionotropic receptors has long been known to cause selective neuronal degeneration (60). Evidence indicates that the delayed form of glutamate-induced neuronal degeneration is mediated by reactive oxygen species (reviewed by Coyle and Puttfarcken [20]). Oxyradicals, depending on their source, can damage cellular proteins, membranes, and DNA, eventually causing cell death. In the case of alcohol withdrawal, increased lipid hydroperoxides may reflect free-radical-induced membrane damage. At the same time, formation of free radicals causes the release of glutamate (32) and inhibits the sodium-dependent high-affinity transport process for glutamate (61), the primary means for removing glutamate from the extracellular space. Thus, a vicious circle of mutually reinforcing processes due to increased glutamatergic neurotransmission and oxidative stress may contribute to neuronal damage in chronic alcoholism. The temporal sequence of the excitatory insult and oxidative stress cannot, however, be determined from these correlative findings but requires additional preclinical studies.
In summary, our data support the hypothesis that augmentation of excitatory synaptic neurotransmission leads to oxidative stress, which in concert with the up-regulation of postsynaptic glutamate receptors, may contribute to the neurotoxicity resulting from long-term alcohol abuse and even from a chronically mild alcohol withdrawal. One possibility suggested by the present data is that the enhanced excitatory neurotransmission not only is present at the acute withdrawal phase but persists for at least 1 month after withdrawal. This persistent effect may be due to a self-perpetuating cycle of enhanced glutamatergic neurotransmission and increased oxidative stress (figure 1). Alternatively, these persistent alterations in excitatory markers in the CSF may represent metabolic traits associated with alcoholism. Another possibility is that chronic alcohol exposure per se augments excitatory neurotransmission and oxidative stress. Although our study could not delineate the time sequence of neuronal events induced by alcohol and did not resolve the trait versus state issue, the results further implicate glutamatergic dysfunction and oxyradicals in the pathophysiology of alcoholism and suggest potential treatments to attenuate neurotoxicity associated with alcohol withdrawal in dependent individuals.
 |
 |
Received July 15, 1997; revision received Nov. 10, 1997; accepted Dec. 22, 1997. From the Laboratory of Molecular and Developmental Neuroscience, Department of Psychiatry, Massachusetts General Hospital and Harvard Medical School, Boston, and the Laboratory of Clinical Studies, Division of Intramural Clinical and Biological Research, National Institute on Alcohol Abuse and Alcoholism, Bethesda, Md. Address reprint requests to Dr. Tsai, Laboratory of Molecular and Developmental Neuroscience, Department of Psychiatry, MGH-CNY, Rm. 2504, 149 13th St., Charlestown, MA 02129; [email protected] (e-mail).
Supported in part by grant NS-13584 from the National Institute of Neurological and Communicative Disorders and Stroke, a Senior Investigator Award to Dr. Coyle from the National Alliance for Research on Schizophrenia and Depression, and funding for Dr. Tsai from the National Alliance for Research on Schizophrenia and Depression (Young Investigator Award), Canavan Foundation, Theodore and Vada Stanley Foundation, and Peter and Elizabeth C. Tower Foundation. Dr. Linnoila died in March 1998.

FIGURE 1. Model of Reduced Inhibitory Neurotransmission and Increased Excitatory Neurotransmission and Oxidative Stress During Alcohol Withdrawal
1 Goodman LS, Gilman A: The aliphatic alcohols, in The Pharmacological Basis of Therapeutics. New York, Macmillan, 1958, pp 98–122Google Scholar
2 Freed WJ, Michaelis EK: Glutamic acid and ethanol dependence. Pharmacol Biochem Behav 1978; 8:509–514Crossref, Medline, Google Scholar
3 Lovinger DM, White G, Weight FF: Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989; 243:1721–1724Crossref, Medline, Google Scholar
4 Blitzer RD, Gil O, Landau EM: Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res 1990; 537:203–208Crossref, Medline, Google Scholar
5 Lustig HS, Chan J, Greenberg DA: Ethanol inhibits excitotoxicity in cerebral cortical cultures. Neurosci Lett 1992; 135:259–261Crossref, Medline, Google Scholar
6 Morrisett RA, Swartzwelder HS: Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci 1993; 13:2264–2272Crossref, Medline, Google Scholar
7 Sinclair JG, Lo GF: Ethanol blocks tetanic and calcium-induced long-term potentiation in the hippocampal slice. Gen Pharmacol 1986; 17:231–233Crossref, Medline, Google Scholar
8 Tsai G, Gastfriend DR, Coyle JT: The glutamatergic basis of human alcoholism. Am J Psychiatry 1995; 152:332–340Link, Google Scholar
9 Weight FF, Aguayo LG, White G, Lovinger DM, Peoples RW: GABA- and glutamate-gated ion channels as molecular sites of alcohol and anesthetic action, in GABAergic Synaptic Transmission: Molecular, Pharmacological, and Clinical Aspects: Advances in Biochemical Psychopharmacology, vol 47. Edited by Biggio G, Concas A, Costa E. New York, Raven Press, 1992, pp 335–347Google Scholar
10 Grant KA, Valverius P, Hudspith M, Tabakoff B: Ethanol withdrawal seizures and the NMDA receptor complex. Eur J Pharmacol 1990; 176:289–296Crossref, Medline, Google Scholar
11 Iorio KR, Reinlib L, Tabakoff B, Hoffman PL: Chronic exposure to cerebellar granule cells to ethanol results in increased N-methyl-D-aspartate receptor function. Mol Pharmacol 1992; 41:1142–1148Medline, Google Scholar
12 Snell LD, Tabakoff B, Hoffman PL: Radioligand binding to the N-methyl-D-aspartate receptor/ionophore complex: alterations by ethanol in vitro and by chronic in vivo ethanol ingestion. Brain Res 1993; 602:91–98Crossref, Medline, Google Scholar
13 Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Hainemann SF, Durman RS, Nestler EJ: Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem 1994; 62:1635–1638Crossref, Medline, Google Scholar
14 Hoffman PL, Grant KA, Snell LD, Reinlib L, Iorio K: NMDA receptors: role in ethanol withdrawal seizures. Ann NY Acad Sci 1992; 654:52–69Crossref, Medline, Google Scholar
15 Sharma AC, Thorat SN, Nayar U, Kulkarni SK: Dizocilpine, ketamine and ethanol reverse NMDA-induced EEG changes and convulsions in rats and mice. Indian J Physiol Pharmacol 1991; 35:111–116Medline, Google Scholar
16 Morgan PF, Nadi NS, Karanian J, Linnoila M: Mapping rat brain structures activated during ethanol withdrawal: role of glutamate and NMDA receptors. Eur J Pharmacol 1992; 225:217–223Crossref, Medline, Google Scholar
17 Sepulveda C, Bustos G, Gysling K, Seguel M, Labarca R: Effects of in vitro ethanol and chronic ethanol consumption on the release of excitatory amino acids in the rat hippocampus. Brain Res 1995; 674:104–106Crossref, Medline, Google Scholar
18 Spanagel R, Zieglgansberger W: Anti-craving compounds for ethanol: new pharmacological tools to study addictive processes. Trends Pharmacol Sci 1997; 18:54–59Crossref, Medline, Google Scholar
19 Sass H, Soyka M, Mann K, Zieglgansberger W: Relapse prevention by acamprosate: results from a placebo-controlled study on alcohol dependence. Arch Gen Psychiatry 1996; 53:673–680; correction, 53:1097Crossref, Medline, Google Scholar
20 Coyle JT, Puttfarcken P: Oxidative stress, glutamate, and neuro~degenerative disorders. Science 1993; 262:689–695Crossref, Medline, Google Scholar
21 Mayfield D, McLeod G, Hall P: The CAGE questionnaire: validations of a new alcoholism screening instrument. Am J Psychiatry 1974; 131:1121–1123Abstract, Google Scholar
22 Selzer M: The Michigan Alcoholism Screening Test: the quest for a new diagnostic instrument. Am J Psychiatry 1971; 127:1653–1658Link, Google Scholar
23 Naranjo CA, Sellers EM: Clinical assessment and pharmaco~therapy of the alcohol withdrawal syndrome. Recent Dev Alcohol 1986; 4:265–281Crossref, Medline, Google Scholar
24 Jones BN: Amino acid analysis by o-phthaldialdehyde precolumn derivatization and reverse-phase HPLC, in Method of Protein Microcharacterization. Edited by Shively JE. Clifton, NJ, Humana Press, 1986, pp 121–151Google Scholar
25 Koller K, Zaczek R, Coyle JT: N-Acetyl-aspartyl glutamate: regional levels in rat brain and the effects of brain lesions as determined by a new HPLC method. J Neurochem 1984; 43:1136–1142Crossref, Medline, Google Scholar
26 Flohe L, Otting F: Superoxide dismutase assays. Methods Enzymol 1984; 105:93–104Crossref, Medline, Google Scholar
27 Puttfarcken PS, Getz RL, Coyle JT: Kainic acid-induced lipid peroxidation: protection with butylated hydroxytoluene and U78517F in primary cultures of cerebellar granular cells. Brain Res 1993; 624:223–232Crossref, Medline, Google Scholar
28 Vevine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz A-G, Ahn BW, Shaltiel S, Stadtman ER: Determination of carbonyl content in oxidatively modified protein, in Methods in Enzymology. Edited by Packer L, Glazer AN. Orlando, Fla, Academic Press, 1990, pp 464–478Google Scholar
29 Foster AC, Kemp JA: Glycine maintains excitement. Nature 1989; 338:377–378Crossref, Medline, Google Scholar
30 Aprison MH: The discovery of the neurotransmitter role of glycine, in Glycine Transmission. Edited by Ottersen OP, Storm-Mathisen J. New York, John Wiley & Sons, 1990, pp 1–24Google Scholar
31 Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch B, Coyle JT: Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol 1990; 28:18–25Crossref, Medline, Google Scholar
32 Pellegrini-Giampietro DE, Cherici G, Alesiani M, Carla V, Mo~roni F: Excitatory amino acid release from rat hippocampal slices as a consequence of free-radical formation. J Neurochem 1988; 51:1960–1963Crossref, Medline, Google Scholar
33 Roy A, DeJong J, Ferraro T, Adinoff B, Ravitz B, Linnoila M: CSF γ-aminobutyric acid in alcoholics and control subjects. Am J Psychiatry 1990; 147:1294–1296Link, Google Scholar
34 Klein RL, Mascia MP, Whiting PJ, Harris RA: GABAa receptor function and binding in stably transfected cells: chronic ethanol treatment. Alcohol Clin Exp Res 1995; 19:1338–1344Crossref, Medline, Google Scholar
35 Freund G, Anderson KJ: Glutamate receptors in the frontal cortex of alcoholics. Alcohol Clin Exp Res 1996; 20:1165–1172Crossref, Medline, Google Scholar
36 Wood PL: The co-agonist concept: is the NMDA-associated glycine receptor saturated in vivo? Life Sci 1995; 57:301–310Google Scholar
37 McBride WJ, Murphy JM, Lumeng L, Li T-K: Effects of ethanol on monoamine and amino acid release from cerebral cortical slices of the alcohol-preferring P line of rats. Alcohol Clin Exp Res 1986; 10:205–208Crossref, Medline, Google Scholar
38 Valverius P, Crabbe JC, Hoffman PL, Tabakoff B: NMDA receptors in mice bred to be prone or resistant to ethanol withdrawal seizures. Eur J Pharmacol 1990; 184:185–189Crossref, Medline, Google Scholar
39 Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW: NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 1996; 16:2034–2043Crossref, Medline, Google Scholar
40 Puttfarcken PS, Handen JS, Montgomery DT, Coyle JT, Werling LL: N-Acetyl-aspartylglutamate modulation of N-methyl-D-as~partate stimulated [3H]norepinephrine release from rat hippocampal slices. J Pharmacol Exp Ther 1993; 266:796–803Medline, Google Scholar
41 Sekiguchi M, Okamoto K, Sakai Y: Excitatory action of N-acetylaspartylglutamate on Purkinje cells in guinea pig cerebellar slices: an intrasomatic study. Brain Res 1987; 423:23–33Crossref, Medline, Google Scholar
42 Sekiguchi M, Okamoto K, Sakai Y: Low concentration N-acetylaspartylglutamate suppresses the climbing fiber response of Purkinje cells in Guinea pig cerebellar slices and the responses to excitatory amino acids of Xenopus laevis oocytes injected with cerebellar mRNA. Brain Res 1989; 482:87–96Crossref, Medline, Google Scholar
43 Sekiguchi M, Wada K, Wenthold RJ: N-Acetylaspartylglutamate acts as an agonist upon homomeric NMDA receptor (NMDAR1) expressed in Xenopus oocytes. FEBS Lett 1992; 311:285–289Crossref, Medline, Google Scholar
44 Slusher BS, Robinson MB, Tsai G, Simmons ML, Richards SS, Coyle JT: Rat brain N-acetylated alpha-linked acidic dipeptidase activity: purification and immunologic characterization. J Biol Chem 1990; 265:21297–21301Medline, Google Scholar
45 Wroblewska B, Wroblewski JT, Saab OH, Neale JH: N-Acetyl~aspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem 1993; 61:943–948Crossref, Medline, Google Scholar
46 Forloni G, Grzanna R, Blakely RD, Coyle JT: Co-localization of N-acetyl-aspartyl-glutamate in central cholinergic, noradrenergic, and serotonergic neurons. Synapse 1987; 1:455–460Crossref, Medline, Google Scholar
47 Tsai G, Coyle JT: N-Acetylaspartate in neuropsychiatric disorder. Prog Neurobiol 1995; 46:531–540Crossref, Medline, Google Scholar
48 Videla LA, Valenzuela A: Alcohol ingestion, liver glutathione and lipoperoxidation: metabolic interrelations and pathological implications. Life Sci 1982; 31:2395–2407Crossref, Medline, Google Scholar
49 Ahmad FF, Cowan DL, Sun AY: Potentiation of ethanol-induced lipid peroxidation of biological membranes by vitamin C. Life Sci 1988; 43:1169–1176Crossref, Medline, Google Scholar
50 Dicker E, Cederbaum AI: Increased NADH-dependent production of reactive intermediates by microsomes after chronic ethanol consumption: comparisons with NADPH. Arch Biochem Biophys 1992; 293:274–280Crossref, Medline, Google Scholar
51 Klein SM, Cohen G, Lieber CS, Cederbaum AI: Increased microsomal oxidation of hydroxyl radical scavenging agents and ethanol after chronic consumption of ethanol. Arch Biochem Biophys 1983; 223:425–432Crossref, Medline, Google Scholar
52 Knecht KT, Bradford BU, Mason RP, Thurman RG: In vivo formation of a free radical metabolite of ethanol. Mol Pharmacol 1990; 38:26–30Medline, Google Scholar
53 Albano E, Tomasi A, Persson J-O, Terelius Y, Goria-Gatti L, Ingelman-Sundberg M, Dianzani MU: Role of ethanol-inducible cytochrome P450 (P450IIE1) in catalysing the free radical activation of aliphatic alcohols. Biochem Pharmacol 1991; 41:1895–1902Crossref, Medline, Google Scholar
54 Bondy S, Guo SX: Effect of ethanol treatment on indices of cumulative oxidative stress. Eur J Pharmacol 1994; 270:349–355Medline, Google Scholar
55 Halliwell B: Reactive oxygen species and the central nervous system. J Neurochem 1992; 59:1609–1623Crossref, Medline, Google Scholar
56 Davis WL, Crawford LA, Cooper OJ, Farmer GR, Thomas DL, Freeman BL: Ethanol induces the generation of reactive free radicals by neural crest cells in vitro. J Craniofac Genet Dev Biol 1990; 10:277–293Medline, Google Scholar
57 Zidenberg-Cherr S, Halsted CH, Olin KL, Reisenauer AM, Keen CL: The effect of chronic alcohol ingestion on free radical defense in the miniature pig. J Nutr 1990; 120:213–217Crossref, Medline, Google Scholar
58 Bondy S, Pearson KR: Ethanol-induced oxidative stress and nutritional status. Alcohol Clin Exp Res 1993; 17:651–654Crossref, Medline, Google Scholar
59 Coudray C, Richard MJ, Faure H, Favier A: Blood and liver lipid peroxide status after chronic ethanol administration in rats. Clin Chim Acta 1993; 219:35–45Crossref, Medline, Google Scholar
60 Olney JW: Excitotoxic amino acids and neuropsychiatric disorders. Annu Rev Pharmacol Toxicol 1990; 30:47–71Crossref, Medline, Google Scholar
61 Volterra A, Trotti D, Tromba C, Floridi S, Racagni G: Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci 1994; 14:2924–2932Crossref, Medline, Google Scholar

