
Huntington’s Disease: Phenomenological Diversity of a Neuropsychiatric Condition That Challenges Traditional Concepts in Neurology and Psychiatry
Huntington’s disease is an autosomal-dominant inherited disorder with full penetrance that is characterized by a triad of motor, cognitive, and psychiatric disturbances. In 1993, its disease etiology was successfully identified as an excess of CAG nucleotide repeats in the IT15 gene on the short arm of chromosome 4 (>38) (1). Because of this polyglutamine expansion, a mutant cytoplasm protein (“huntingtin”) of presently unknown function is expressed and is possibly responsible for neurotrophic deficits, abnormal vesicle fusion, and amyloid-like inclusions in striatal nerve cells (2, 3). Depending on the individual CAG expansion load, disease onset generally occurs between the ages of 30 and 40, entailing an invariably lethal progression within 10 to 20 years (4). Characteristic neuropathological features include the degeneration of the head of the caudate nucleus and other striatal areas as well as frontally pronounced cortical atrophy without temporal lobe involvement (5). Aside from new treatment approaches that examine the benefit of fetal striatal transplantation, current disease management is still entirely symptomatic (6).
Neurodegenerative alterations and its presentation as a movement disorder account for the traditional linkage of Huntington’s disease to the field of neurology. Within this context, the divergent development of psychiatric and neurologic disciplines during the last century (“the great divide” [7]) may explain the common neglect of psychiatric manifestations in Huntington’s disease patients (8). Recent advances of clinical neuroscience, however, challenge the obsolete mind-body dichotomy with concepts emphasizing the plasticity of brain structure in mediating behavioral and emotional (dys)function (9). By adapting a modern holistic approach, the following case report emphasizes the spectrum and clinical significance of neuropsychiatric phenomena associated with Huntington’s disease. Diagnostic difficulties are exemplified and discussed in light of the traditional separation of neurological and psychiatric facets in the clinical setting (7). Within this context, the current scientific knowledge of Huntington’s disease’s pleiotropy is reviewed along with a discussion of the diagnostic power of specific neuropsychological approaches.
Case History
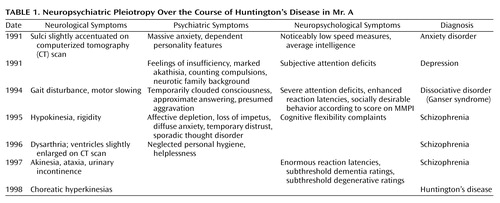
The present report describes the case of Mr. A, a 49-year-old man with rapidly increasing physical and mental impairments who had been diagnosed with Huntington’s disease 1 year before admission to our department. Analysis of the patient’s medical history revealed a long history of various neuropsychiatric disturbances, diagnoses, and treatment approaches preceding the differential diagnosis and genetic verification of the disease (Table 1).
Personal and Social Background
Mr. A was born in 1950 after an uncomplicated pregnancy, labor, and delivery. His natural father had abandoned the patient’s family and three other siblings when he had been only 3 years old. According to his mother, he was an anxious and bed-wetting child who was often the target of his stepfather’s violent outbursts. At the age of 12, he exhibited recurrent cataplectic attacks with a sudden loss of muscle tone and immobility accompanied by unaltered consciousness. Although he was actually reported to be an intelligent and successful student, the frequency of the attacks disabled the boy so much that he had to repeat a school year. The results of a standard neurological examination yielded no obvious irregularities, so the symptoms were attributed to an “unknown psychological causation.” Along with his remarkable family background, this diagnosis may have misguided the subsequent appraisals of his therapists. At age 20, after successful completion of secondary school and training as an insurance agent, Mr. A got married. According to his wife, he exhibited dependent and jealous behavior right from the beginning of their relationship. Apart from this, premorbid social and occupational functioning was always unobtrusive. During 1990, our patient experienced various phobic symptoms of increasing severity, leading to prolonged sick leave and inpatient treatment after acute exacerbation of the symptoms and separation from his spouse. As a result of the unknown fate of the patient’s biological father, his family history remained incomplete. None of Mr. A’s available relatives was afflicted with a neuropsychiatric disorder.
Neuropsychiatric Development
At the time of his first admission in 1991, Mr. A showed signs of intense fear, including tachycardia, hyperhydrosis, and extreme stuttering, which prevented any reasonable exploration before the application of benzodiazepines. He reported unspecific “worryness” and recurrent states of massive anxiety, which were generally but not exclusively situation-bound (e.g., in darkness, crowded rooms, or while driving a car). No clear psychotic symptoms were seen at that time.
Initial Diagnosis: Neurotic Anxiety
The clinical examination of Mr. A’s neurological and electroencephalographic status yielded no pathological findings, a computerized tomography (CT) scan equally failed to reveal any striking irregularities. In addition to an average IQ level, his neuropsychological screening revealed varied results regarding Mr. A’s attentional functions. This inhomogeneous test profile was obviously caused by inferior test performance in all speed-dependent subscales (>1.5 SDs below mean performance). Unfortunately, this pathognomic indicator was not traced further. Instead, the therapist’s attention was attracted by the “obvious childhood origin of the neurotic symptomatology,” and a neurotic anxiety disorder was subsequently diagnosed (DSM-III-R, code 300.0). The therapeutic regime consisted of a β-blocking intervention with propranolol hydrochloride and referral for outpatient behavioral therapy. In the absence of clear psychotic symptoms, further progression of the clinical picture was characterized by various psychiatric symptoms exacerbating as a result of environmental stressors, a gradual increase of subtle cognitive and psychomotor constraints, and growing dependence concerning daily demands.
Second Diagnosis: Endogenous Depression
Approximately 4 weeks later, Mr. A had to be readmitted in a desolate state after he had started to sleep on park benches after a severe dispute with his mother. Retrospectively, Mr. A reported a broad array of psychiatric symptoms, including feelings of insufficiency, helplessness, and pronounced attentional deficits (Table 1). Intense psychomotor restlessness prevented him from sitting down during meals or conversation. For understandable reasons, Mr. A’s thoughts were narrowed to worries concerning his medical problems and the painful separation efforts of his wife. Unfortunately, the professional’s view of that time remained overly focused on Mr. A’s “extensive neurotic family background,” and his worries were interpreted as “pronounced hypochondriac ideas.” At this time, his complaints were classified as endogenous depression (DSM-III-R, code 296.2) and treated thymoleptically with amitriptyline.
Third Diagnosis: Ganser Syndrome
In December 1994, Mr. A was admitted to the hospital with a presumed psychotic disorder of organic origin. As a result of the continuous quarrel with his relatives, he had become homeless and repeatedly spent the night in his car. On admission he was disoriented, exhibited a partially clouded consciousness, psychomotor slowing, and a seesaw gait disturbance. During the clinical interview, the psychiatrist adopted previous psychodynamic explanations and focused on the pronounced approximate answers of the patient, especially when his marital relationship was addressed. More precisely, the lack of insight concerning separation efforts by his wife left the impression of an intentional aggravation of his complaints in terms of a maladjusted defense style. This interpretation was supported by the psychological disclosure of a tendency to pattern his behavior according to its social desirability (MMPI). Subsequently, a dissociative disorder specified as “Ganser syndrome” (DSM-III-R, code 300.15) was diagnosed, and the basic necessity for a long-term psychotherapeutic confrontation with his adaptive difficulties was advised.
Fourth Diagnosis: Schizophrenia
Four weeks later, Mr. A was hospitalized in yet another state mental institution with pronounced affective depletion, substantial loss of impetus, and psychomotor slowing interpreted as residual negative symptoms. For the next 2 years, he remained hospitalized with a diagnosis of schizophrenia, the fluctuating clinical picture scarcely stabilized with clozapine treatment (DSM-III-R, code 295.2; DSM-IV, code 295.6). His complaints consisted of diffuse anxiety symptoms, reduced attentional capacities and mental flexibility, as well as increasing hypokinesia and rigidity. Positive psychotic symptoms were present to a lesser extent and consisted of intermittent stages of pronounced distrust and formal thought disorder. In 1996, the patient’s status showed further deterioration, since he had now also begun to neglect his personal hygiene, needed support in managing even trivial demands, and developed dysarthric speech.
Final Diagnosis: Huntington’s Disease
One year later, the clinical picture also included akinesia, ataxia, and urinary incontinence. Even then, his results on conventional neuropsychological tests focused on the assessment of dementia (cognitive section of the Cambridge Examination for Mental Disorders of the Elderly, the Mini-Mental Status Examination [MMSE], the Structured Interview for the Diagnosis of Dementia) and degenerative brain processes were not clearly in the pathological range (Cambridge Examination for Mental Disorders of the Elderly, total score=87; MMSE, total score=26). Genetic testing for Huntington’s disease was not considered until clear choreatic hyperkinesias emerged 7 years after his first psychiatric referral. Despite the patient’s widespread mental and physical disability, standard neurological, neuroradiological, and neuropsychological procedures still provided no reliable evidence for a primary neurodegenerative disease. The origin of the genetic defect was not definitely determinable from the available evidence. Because of the early disappearance of the patient’s father, a de novo expansion of the unaffected parents’ CAG was just as plausible as a paternal transmission of the CAG expansion.
Neuropsychiatric Profile at Current Admission
Mr. A’s last admission to our institute had been initiated by the local police. Now a resident in a rehabilitative institution for psychiatric patients, he was found in a streetcar, mute, petrified, and trembling, unable to give his identity on request. Clinical observations confirmed the initial impression of a further deterioration of his condition. To define his remaining functional capacities more precisely, a detailed psychiatric, neurological, and neuropsychological assessment was carried out.
Psychiatric and Neurological Examination
In addition to previously known cognitive deficits, Mr. A now exhibited a marked gait and balance disturbance, often requiring the use of a wheelchair. Psychopathologically, an overall reduction of apprehension, impetus, affective expression, and attention was concurrent with a tendency for social withdrawal. Although conversation was notably complicated by pronounced dysarthria, he comprehensively reported subjective distress because of his urinary incontinence. Furthermore, a recently emerged swallowing disability had demanded recurrent pulp nourishment and had given rise to persistent asphyxiation fears while he was eating. In contrast to the pronounced slowing of intentional motor functions, choreatic hyperkinesias of the face and trunk were only sporadically observable. Overall, the clinical impression of end-stage Huntington’s disease arose. Therapeutic interventions focused on pharmacological stabilization with clozapine, tiapride, and tetrabenazine.
Neuroimaging
Brain imaging was performed by using standard proton-density-weighted, fluid-attenuated inversion recovery and three-dimensional volumetric magnetic resonance sequences. In contrast to previous CT scans, these morphological pictures verified the characteristic neurodegenerative changes of Huntington’s disease (Figure 1). Because of the considerable atrophy of the head of the caudate nucleus, the frontal horns of the lateral ventricles appeared bilaterally enlarged. An additional target of neuronal loss was localized in the putamen. In contrast to the focal neuronal loss of the striatum, the widespread accentuation of cortical sulci is in line with diffuse cerebral atrophy.
Neuropsychological Findings
Considering the known dementia syndrome secondary to Huntington’s disease, the current cognitive examination focused on the assessment of Mr. A’s mnestic functions. In line with the psychological evaluation 2 years earlier, we performed a structured interview for the diagnosis of dementia of the Alzheimer type, multi-infarct dementia, and dementias of other etiology (Structured Interview for the Diagnosis of Dementia), the Cambridge Examination for Mental Disorders of the Elderly, as well as the MMSE. Problem-solving and executive capabilities were analyzed with the Wisconsin Card Sorting Test (10). Behavioral, functional, and motor abnormalities were estimated by using the Unified Huntington’s Disease Rating Scale (11). Furthermore, a broad range of basic attentional functions were examined by using the computerized Test Battery for the Assessment of Attention (12).
In accordance with the clinical impression, the Unified Huntington’s Disease Rating Scale motor examination revealed a moderate movement disability, with pronounced signs of bradykinesia, dysarthria, and coordination deficits (total score=25). The behavioral assessment disclosed mild depressive symptoms (total score=8). In contrast to our initial expectations, the applied dementia ratings again did not indicate the presence of a dementia syndrome. Although a slight decline in nearly all subscales was observable compared to the prior evaluation, the various dementia ratings still did not grasp the obvious cognitive constraints of our patient (Cambridge Examination for Mental Disorders of the Elderly, total score=80; MMSE, total score=24).
However, the evaluation of our patient’s basic attentional capabilities (Test Battery for the Assessment of Attention) and executive functions (Wisconsin Card Sorting Test) clearly pointed to the presence of pronounced cognitive and psychomotor deficits. Although Mr. A showed regular reactions to task-contingent external stimuli (phasic alertness value: 76th percentile), marked psychomotor slowing was obvious while we determined his basic reaction speed (his median reaction time was less than the first percentile). His attempts to handle the working memory and response flexibility tasks resulted in a complete performance breakdown, indicating an exceptional dorsolateral-prefrontal processing deficit (omission responses were false alarms of less than the first percentile). The consistently low Wisconsin Card Sorting Test performance scores also revealed apparent difficulties regarding frontally mediated skills, especially cognitive flexibility and abstract reasoning (total categories and perseverations scores: less than the first percentile). In contrast, the arousal-related internal maintenance of attention in the vigilance task was unaffected (omissions and false alarms: 42 responses, 76th percentile). Overall, the neuropsychological profile of Mr. A pointed to a locally constricted frontal processing deficit affecting prefrontal cognitive and motor areas rather than the midtemporal-hippocampal regions usually associated with neurodegenerative memory complaints.
Discussion
Phenomenological Diversity of Initial Huntington’s Disease
Although a cure for Huntington’s disease is still not available, differential diagnosis options have expanded enormously through precise characterization of the genetic locus in 1993. As a result, the identification and examination of presymptomatic gene carriers and early disease stages widened the known spectrum of characteristic clinical symptoms in motor, cognitive, and psychiatric domains (13). Within a considerable range, the underlying genetic defect consisting of variable CAG nucleotide expansions seems to be related to disease onset and symptom severity and is thus prognostically valuable (14). In contrast to other genetic disease modes, the variability of triplet-repeats points to a neurobiological and behavioral spectrum disorder associated with individual genetic load. The complex interaction of this particular genetic defect with innumerable environmental factors and genes implies a broad array of different possible phenotypes among patients with the same disorder. From this point of view, the traditionally emphasized signs of Huntington’s disease, “chorea plus dementia,” may be inadequately narrowed core features for diagnostic guidance in the early stages of the disease (14).
Spectrum of Motor Abnormalities
Descriptions of Huntington’s disease motor signs focusing on choreatic hyperkinesias can be traced back to George Huntington’s original work in 1872 (15). As a sudden and intermittent loss of involuntary motor control, chorea manifests itself as complex, rapid, arrhythmic, and irregular movements associated with muscular hypotonus. In accordance with the known significance of the basal ganglia for involuntary as well as voluntary motor actions, the modern empirical literature emphasizes the broad array of Huntington’s disease motor disturbances. Within this context, chorea represents only a small part of the phenomenological spectrum (14). Nonchoreatic motor abnormalities, possibly as a consequence of faulty error feedback control (16), are not restricted to symptomatic stages of the disease but are already observable in asymptomatic gene carriers (17). In a longitudinal study, Kirkwood and co-workers (18) confirmed the development and progression of various motor disturbances in gene carriers subthreshold to clinical diagnosis (18). Daily functioning may be impaired by coexisting problems concerning the initiation and execution of voluntary movements, oculomotor deficits, akathisia, and particularly bradykinesia (18, 19).
Mirroring the clinical picture of our patient, the motor phenomenology of later stages appears akinetic-rigid, with overall slowing of intentional motor actions, gait abnormalities, dysarthria, and dysphagia (20). Additionally, urinary incontinence may develop as a result of choreatic contractions of abdominal perineal floor muscles (21, 22). Prognostically relevant, bradykinesia seems to be strongly associated with disease stage and characteristic loss of striatal D2 binding, while evidence for a reliable prediction of Huntington’s disease development by subtle involuntary movements is lacking (20).
Development of Cognitive Deficits
Dementia, the progressive decline of cognitive functions “amounting to insanity” (15), resembles another traditional core feature of the clinical picture originally noted by George Huntington. According to the fundamental descriptions by Brandt and Bylsma (23), the dementia of Huntington’s disease comprises an initial reduction of complex abilities (memory, planning) as well as basic cognitive functions (attention) causing performance breakdown in cognitive screenings (e.g., MMSE) and dementia rating scales. Extensive states of mental deterioration, however, seem to be restricted to advanced stages of the disease (24).
Along with a greater understanding of the functional properties of striatothalamocortical processing loops, the cognitive impact of the basal ganglia and other subcortical structures has been extensively discussed (for a review, see reference 25). Considering the massive striatal input to the frontal cortex, it is not surprising that there is good empirical evidence indicating Huntington’s disease-associated deficits in executive functioning and IQ performance subscales similar to those found in our patient (26, 27).
Although not always consistent, current scientific knowledge points to a loss of striatal neurons before the manifestation of clinical symptoms (28). In line with these results, Paulsen et al. (29) and other groups note the emergence of subtle cognitive impairments years before the appearance of overt motor signs. Within this context, these and other authors emphasize the necessity of applying scientifically well-established and sensitive neuropsychological measures that indicate the “phenoconversion” of gene carriers from an asymptomatic to a symptomatic state (28).
Sprengelmeyer and co-workers (30) stimulated the neuropsychological research on Huntington’s disease with the notion that higher-order cognitive problems may, in fact, be the consequence of antecedent, basic functional deficits. The particular pattern of attentional deficits described in this context noticeably resembles the neuropsychological profile of our patient. Fundamental abilities, such as response inhibition, divided attention, and response flexibility, are severely affected, while externally triggered arousal functions seem to be unimpaired. Bradykinesia—causing extreme reaction latencies in neuropsychological test settings—is frequently noted as a characteristic aspect of Huntington’s disease performance even in asymptomatic gene carriers (30, 31). Furthermore, basic speed-dependent measures without elaborate cognitive demands are reported to be superior in monitoring disease progression and seem to be associated with the individual CAG expansion (32). Other preclinical impairments may manifest themselves during attention shifting, verbal fluency, learning, and visuospatial functions, with inconsistent data being available on the correlation of these symptoms with the individual genetic loading (33).
Early Psychiatric Manifestations
Although the majority of patients and their caregivers are substantially impaired by neuropsychiatric disturbances associated with Huntington’s disease, this aspect of the Huntington’s disease symptom triad has received comparatively little attention (8, 34). After reviewing the current literature, we must draw the conclusion that there may be a wide range of concomitant psychiatric complications. A recent study (8) found that 98% of the examined Huntington’s disease patients were afflicted with unspecific neuropsychiatric problems, especially dysphoria, agitation, irritability, apathy, and anxiety (8). Other authors additionally report a disproportionately high prevalence of obsessive-compulsive symptoms, sexual disorders, sleep disturbances, explosive behaviors, personality changes, psychotic symptoms, and suicidal tendencies (35, 36).
Our case history documents the clinical significance, heterogeneity, and diagnostic difficulties associated with these neuropsychiatric complaints. In clinical settings, physicians may be confronted with only one of these signs as the sole presenting feature of the picture of Huntington’s disease, possibly preceding the occurrence of more familiar symptoms by up to 20 years (37). The situation is further complicated by the fact that the amount of symptoms exhibited independent of the Huntington’s disease genotype cannot be determined by the therapist, a factor that questions the predictive power of molecular genetics in current neuropsychiatry. Additional problems of disease management arise from the merging of neurodegenerative and reactive processes or the pharmacological provocation of masked delirious states, as rightfully pointed out in a recent publication (38). It is possible that some of the first psychiatric symptoms of our patient, such as general anxiety, were unrelated to Huntington’s disease and indeed caused by negative social interactions independent of Huntington’s disease. However, the Huntington’s disease-related deficit in speed-related cognitive abilities may already have interfered with the patient’s social coping abilities.
Implications: An Extended Neuropsychiatric Perspective
Modern neuroscience permits a holistic understanding of neuronal functioning, thereby integrating antiquated dichotomies of “mind versus body” or “gene versus environment.” In conceptualizing health as well as sickness in terms of a “biopsychological phenomenon” (39), continued efforts in brain research may lead to a new nosology of neurological and mental diseases (40). Current models of brain circuitry—e.g., the open interconnected model of striatothalamocortical projections by Joel (41)—allow a deeper understanding of the full range of neuropsychiatric symptoms associated with the Huntington’s disease genotype. Furthermore, neuroscience may succeed in overcoming the traditional clinical, scientific, and educational segregation of neurology versus psychiatry, a development that has already been anticipated and discussed as the “closing of a great divide” in an important publication by Price et al. (7).
Even to date, the enormous phenotypic heterogeneity and the occurrence of sporadic cases provoke a substantial diagnostic inaccuracy in Huntington’s disease (42). Considering the exceptional pleiotropy and the unusual family history of the present case, any retrospective conclusions would certainly be of only limited use. Nevertheless, the initial unspecific clinical presentation and further progression of our case shows a striking overlap with the scientific knowledge of Huntington’s disease phenomenology. In our opinion, the unfortunate course of the present clinical history may be linked to the insufficient bridging of this “great divide.” From a neuropsychiatric point of view, the range of psychiatric and neuropsychological symptoms presented by our patient may have been underestimated. Although the deficits of voluntary motor performance associated with Huntington’s disease had accompanied disease progression for years, genetic testing was not considered until highly visible symptoms of chorea emerged. Once identified as a neurodegenerative movement disorder, psychotherapeutic interventions were abruptly discontinued, although a recent publication indicates their effectiveness in a multimodal treatment approach (38).
In light of the described neurotic family background, psychiatric evaluations may have likewise been narrow-minded concerning the potential neurodegenerative impact of the noticed behavioral and emotional disturbances. Even classical symptoms, such as our patient’s approximate answering (“Ganser syndrome”), the hypochondriac ideas, and his stubborn appraisal of a happy marital relationship (“reduced insight”) may reflect initial deficits in cognitive flexibility and executive planning due to organic causes. Furthermore, the fluid exacerbation of the clinical picture with environmental stressors biased diagnostic considerations so that possible structural correlates were not explored. Finally, the psychological efforts to outline our patient’s initial presentation may have been unduly focused on the assessment of advanced neurodegenerative deficits. Considering the cognitive and motor profile of the onset of Huntington’s disease reported in the current literature, important diagnostic clues were probably missed because of the incomplete consideration of brain functional networks. A further encouragement of a widened neuropsychiatric perspective in clinical settings and the utilization of modern neuropsychological tools will influence disease management to the benefit of our patients (9).
 |
Received Aug. 5, 2002; revision received July 1, 2003; accepted July 11, 2003. From the Nuclear Magnetic Resonance–Research in Psychiatry, Central Institute of Mental Health, University of Heidelberg, Heidelberg, Germany; the NeuroImage Nord and Department of Psychiatry and Psychotherapy, University of Hamburg, Hamburg, Germany; the Department of Psychiatry and Psychotherapy of the Charité, Campus Mitte, Humboldt University of Berlin, Berlin, Germany. Address reprint requests to Dr. Braus, Department of Psychiatry and Psychotherapy, Martinistraße 52, 20246 Hamburg, Germany; [email protected] (e-mail).

Figure 1. Magnetic Resonance Imaging (MRI) Scans of Striatal Degeneration in a Patient With Huntington’s Disease and in a Healthy Comparison Subjecta
aThe images reveal a noticeable enlargement of the anterior portion of the lateral ventricles in the patient due to partial degeneration of the head of the nucleus caudatus.
1. Huntington’s Disease Collaborative Research Group: A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72:971–983Crossref, Medline, Google Scholar
2. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E: Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001; 293:493–498Crossref, Medline, Google Scholar
3. Morton AJ, Faull RL, Edwardson JM: Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res Bull 2001; 56:111–117Crossref, Medline, Google Scholar
4. Kehoe P, Krawczak M, Harper PS, Owen MJ, Jones AL: Age of onset in Huntington disease: sex-specific influence of apolipoprotein E genotype and normal CAG repeat length. J Med Genet 1999; 36:108–111Medline, Google Scholar
5. Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E: Regional specificity of brain atrophy in Huntington’s disease. Exp Neurol 1998; 154:663–672Crossref, Medline, Google Scholar
6. Philpott LM, Kopyov OV, Lee AJ, Jacques S, Duma CM, Caine S, Yang M, Eagle KS: Neuropsychological functioning following fetal striatal transplantation in Huntington’s chorea: three case presentations. Cell Transplant 1997; 6:203–212Crossref, Medline, Google Scholar
7. Price BH, Adams RD, Coyle JT: Neurology and psychiatry: closing the great divide. Neurology 2000; 54:8–14Crossref, Medline, Google Scholar
8. Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL: Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry 2001; 71:310–314Crossref, Medline, Google Scholar
9. Kandel ER: Biology and the future of psychoanalysis: a new intellectual framework for psychiatry revisited. Am J Psychiatry 1999; 156:505–524Abstract, Google Scholar
10. Heaton RK, Chelune GJ, Talley JL, Kay GG, Curtiss G: Wisconsin Card Sorting Test Manual. Odessa, Fla, Psychological Assessment Resources, 1993Google Scholar
11. Huntington Study Group: Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord 1996; 11:136–142Crossref, Medline, Google Scholar
12. Zimmermann P, Fimm B: Testbatterie zur Erfassung von Aufmerksamkeitsstörungen, version 1.02. Freiburg, Germany, Psytest, 1993Google Scholar
13. Squitieri F, Cannella M, Giallonardo P, Maglione V, Mariotti C, Hayden MR: Onset and pre-onset studies to define the Huntington’s disease natural history. Brain Res Bull 2001; 56:233–238Crossref, Medline, Google Scholar
14. Squitieri F, Berardelli A, Nargi E, Castellotti B, Mariotti C, Cannella M, Lavitrano ML, de Grazia U, Gellera C, Ruggieri S: Atypical movement disorders in the early stages of Huntington’s disease: clinical and genetic analysis. Clin Genet 2000; 58:50–56Crossref, Medline, Google Scholar
15. Huntington G: On chorea. Med Surg Rep 1872; 26:320–321Google Scholar
16. Smith MA, Brandt J, Shadmehr R: Motor disorder in Huntington’s disease begins as a dysfunction in error feedback control. Nature 2000; 403:544–549Crossref, Medline, Google Scholar
17. de Boo GM, Tibben A, Lanser JB, Jennekens-Schinkel A, Hermans J, Maat-Kievit A, Roos RA: Early cognitive and motor symptoms in identified carriers of the gene for Huntington disease. Arch Neurol 1997; 54:1353–1355Crossref, Medline, Google Scholar
18. Kirkwood SC, Siemers E, Stout JC, Hodes ME, Conneally PM, Christian JC, Foroud T: Longitudinal cognitive and motor changes among presymptomatic Huntington disease gene carriers. Arch Neurol 1999; 56:563–568Crossref, Medline, Google Scholar
19. Bradshaw JL, Phillips JG, Dennis C, Mattingley JB, Andrewes D, Chiu E, Pierson JM, Bradshaw JA: Initiation and execution of movement sequences in those suffering from and at-risk of developing Huntington’s disease. J Clin Exp Neuropsychol 1992; 14:179–192Crossref, Medline, Google Scholar
20. Sánchez-Pernaute R, Künig G, del Barrio Alba A, de Yébenes JG, Vontobel P, Leenders KL: Bradykinesia in early Huntington’s disease. Neurology 2000; 54:119–125Crossref, Medline, Google Scholar
21. Kirkwood SC, Su JL, Conneally P, Foroud T: Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol 2001; 58:273–278Crossref, Medline, Google Scholar
22. Wheeler JS, Sax DS, Krane RJ, Siroky MB: Vesico-urethral function in Huntington’s chorea. Br J Urol 1985; 57:63–66Crossref, Medline, Google Scholar
23. Brandt J, Bylsma F: The dementia of Huntington’s disease, in the Neuropsychology of Alzheimer’s Disease and Other Dementias. Edited by Parks RW, Zec RF, Wilson RS. New York, Oxford University Press, 1993, p 681Google Scholar
24. Bamford KA, Caine ED, Kido DK, Cox C, Shoulson I: A prospective evaluation of cognitive decline in early Huntington’s disease: functional and radiographic correlates. Neurology 1995; 45:1867–1873Crossref, Medline, Google Scholar
25. Middleton FA, Strick PL: Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev 2000; 31:236–250Crossref, Medline, Google Scholar
26. de Boo GM, Tibben A, Lanser JB, Jennekens-Schinkel A, Hermans J, Vegter-van der Vlis M, Roos RA: Intelligence indices in people with a high/low risk for developing Huntington’s disease. J Med Genet 1997; 34:564–568Crossref, Medline, Google Scholar
27. Foroud T, Siemers E, Kleindorfer D, Bill DJ, Hodes ME, Norton JA, Conneally PM, Christian JC: Cognitive scores in carriers of Huntington’s disease gene compared to noncarriers. Ann Neurol 1995; 37:657–664Crossref, Medline, Google Scholar
28. Aylward EH, Codori AM, Rosenblatt A, Sherr M, Brandt J, Stine OC, Barta PE, Pearlson GD, Ross CA: Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington’s disease. Mov Disord 2000; 15:552–560Crossref, Medline, Google Scholar
29. Paulsen JS, Zhao H, Stout JC, Brinkman RR, Guttman M, Ross CA, Como P, Manning C, Hayden MR, Shoulson I: Clinical markers of early disease in persons near onset of Huntington’s disease. Neurology 2001; 57:658–662Crossref, Medline, Google Scholar
30. Sprengelmeyer R, Lange H, Homberg V: The pattern of attentional deficits in Huntington’s disease. Brain 1995; 118:145–152Crossref, Medline, Google Scholar
31. Siemers E, Foroud T, Bill DJ, Sorbel J, Norton JA Jr, Hodes ME, Niebler G, Conneally PM, Christian JC: Motor changes in presymptomatic Huntington disease gene carriers. Arch Neurol 1996; 53:487–492Crossref, Medline, Google Scholar
32. Snowden J, Craufurd D, Griffiths H, Thompson J, Neary D: Longitudinal evaluation of cognitive disorder in Huntington’s disease. J Int Neuropsychol Soc 2001; 7:33–44Crossref, Medline, Google Scholar
33. Giordani B, Berent S, Boivin MJ, Penney JB, Lehtinen S, Markel DS, Hollingsworth Z, Butterbaugh G, Hichwa RD, Gusella JF, et al: Longitudinal neuropsychological and genetic linkage analysis of persons at risk for Huntington’s disease. Arch Neurol 1995; 52:59–64Crossref, Medline, Google Scholar
34. Naarding P, Kremer HP, Zitman FG: Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. Eur Psychiatry 2001; 16:439–445Crossref, Medline, Google Scholar
35. Cummings JL, Cunningham K: Obsessive-compulsive disorder in Huntington’s disease. Biol Psychiatry 1992; 31:263–270Crossref, Medline, Google Scholar
36. Anderson KE, Louis ED, Stern Y, Marder KS: Cognitive correlates of obsessive and compulsive symptoms in Huntington’s disease. Am J Psychiatry 2001; 158:799–801Link, Google Scholar
37. Folstein S, Abbott MH, Chase GA, Jensen BA, Folstein MF: The association of affective disorder with Huntington’s disease in a case series and in families. Psychol Med 1983; 13:537–542Crossref, Medline, Google Scholar
38. Blass DM, Steinberg M, Leroi I, Lyketsos CG: Successful multimodality treatment of severe behavioral disturbance in a patient with advanced Huntington’s disease (case conf). Am J Psychiatry 2001; 158:1966–1972Link, Google Scholar
39. Merino JG: Neurology and psychiatry: closing the great divide. Neurology 2000; 55:602–603Crossref, Medline, Google Scholar
40. Cowan WM, Harter DH, Kandel ER: The emergence of modern neuroscience: some implications for neurology and psychiatry. Annu Rev Neurosci 2000; 23:343–391Crossref, Medline, Google Scholar
41. Joel D: Open interconnected model of basal ganglia-thalamocortical circuitry and its relevance to the clinical syndrome of Huntington’s disease. Mov Disord 2001; 16:407–423Crossref, Medline, Google Scholar
42. Folstein SE, Leigh RJ, Parhad IM, Folstein MF: The diagnosis of Huntington’s disease. Neurology 1986; 36:1279–1283Crossref, Medline, Google Scholar

