
Identification of the Slynar Gene (AY070435) and Related Brain Expressed Sequences as a Candidate Gene for Susceptibility to Affective Disorders Through Allelic and Haplotypic Association With Bipolar Disorder on Chromosome 12q24
Abstract
Objective: Three linkage studies of bipolar disorder have implicated chromosome 12q24.3, with significant lod scores of over 3.00. Several other linkage studies have found lod scores between 2.00 and 3.00. In order to identify which gene on this chromosome is responsible, the authors carried out tests of allelic association with bipolar disorder in order to fine map an affective disorder susceptibility gene. Method: DNA samples from 681 bipolar disorder patients and 570 comparison subjects from Denmark and the United Kingdom were genotyped with markers close to the region at which the authors had found maximum linkage in previous studies. Results: Single marker allelic association was found with four markers in the Danish cohort. Seven markers in exactly the same region were then found to show significant allelic association in the U.K. cohort. Tests of haplotypic association were also significant, confirming the single marker allelic associations. Conclusions: These positive fine mapping results validate earlier linkage studies and implicate a 278-kilobase region of chromosome 12 that contributes to the etiology of bipolar disorder. Several brain transcripts are transcribed from sequences in the region. The main candidate gene has no known function but is found in human brain cDNA and is homologous to a Macaque brain cDNA. Sequencing of expressed sequences and control regions in the area should identify etiological base pair changes that increase susceptibility to bipolar disorder.
Bipolar affective disorder (BPAD [MIM 125480]) has a lifetime risk of 0.3%–1.5%. The disorder is characterized by episodes of mania and depression. Family, twin, and adoption studies have demonstrated that genetic factors contribute strongly to the etiology of bipolar disorder and genetically related unipolar affective disorders (1 – 3) . However, no precise etiological base pair changes or mutations within or related to any genes have yet been found on any chromosome. A maximum lod score of 2.11 showing linkage between a genetic marker and bipolar disorder was first found on chromosome 12q23-q24.1 near the Darier’s disease gene locus (4) . We subsequently published linkage evidence for bipolar and unipolar affective disorder to chromosome 12q24 with a lod score of 3.37 and with nonparametric linkage p values between 0.005 and 0.00003 (5) . Independent linkage studies have found lod scores of 1.24 in U.S. families (6) , 3.92 and then 5.50 from a French Canadian isolate (7 , 8) , and 2.80 in a cohort of U.K. and Icelandic families (9) . We have also published evidence for significantly increased haplotype sharing and allelic association on 12q24 in bipolar subjects from a genetic isolate in the Faroe Islands (10) . A further linkage study of bipolar disorder with markers near the Darier’s gene locus and a haplotype analysis of previously published pedigrees claimed to have identified a critical region containing a susceptibility locus in a 6.5-Mb region on 12q23-q24 (11) . DNA sequencing and denaturing high-performance liquid chromatography screening of the Darier’s gene in a bipolar family group, a comparison group, and in Darier’s disease subjects could not find any etiological base pair changes in bipolar disorder cases (12 , 13) . Linkage analysis of a large cohort of families multiply affected with unipolar affective disorder has also shown very good evidence for linkage at 12q22-23.2 with a lod score of 4.60 (14) . Two attempts to fine map a bipolar susceptibility locus by detecting association between markers on 12q24 and bipolar disorder have been made. In a Canadian cohort (15) , allelic association was found with the markers labeled NBG6 (p=0.008) and NBG12 (p<0.0001). In the NBG12 area 32 genes were screened for the presence of polymorphisms in coding sequences and intron/exon junctions. Twenty-two nonsynonymous single nucleotide polymorphisms (SNPs) were found and genotyped. Two uncommon polymorphisms were found in the KIAA1595 and FLJ22471 genes, and these were associated with p values below 0.05 with the T1 CLUMP statistic. In a U.K. cohort of 347 bipolar affective disorder patients and 374 comparison subjects (16) , an allelic association (p=0.0016) was identified with one microsatellite marker and two SNPs (rs3847953 and rs933399), and an insertion/deletion with putative functional relevance gave uncorrected p values ranging from p=0.005 to p=0.002 in a 150-Kb region around the CUX2 and FLJ32356 genes on 12q23-q24. We independently set out to follow up the 12q24 linkage findings by fine mapping the exact position of a 12q24 bipolar susceptibility locus in two cohorts originating from Denmark and the United Kingdom.
Method
The U.K. cohort consisted of 600 bipolar disorder patients that included 549 (93%) bipolar I cases with psychotic symptoms and 41 bipolar II cases (7%) according to Research Diagnostic Criteria (RDC) categories. Patients were excluded if there was any evidence of a “secondary” or “symptomatic” cause of bipolar disorder. All the bipolar II disorder patients had received inpatient or outpatient psychiatric treatment. The comparison subjects were 450 screened normal volunteers. The patients and comparison subjects were selected if both their parents and all four grandparents were of Irish, Welsh, Scottish, or English ancestry as defined by an ancestry checklist. Subjects were also included if one of the four grandparents was of European ancestry before the 2004 EU enlargement. U.K. National Health Service (NHS) multicenter and local research ethics committee approval was obtained, and all subjects signed an approved consent form after reading an information sheet and after a complete description of the study was given to each subject. All subjects were interviewed by a psychiatrist who used the lifetime version of the Schizophrenia and Affective Disorders Schedule. All the bipolar patients were also rated with the 90-item OPCRIT checklist (17) . Diagnoses were assigned using Research Diagnostic Criteria. The Danish cohort consisted of 81 bipolar I disorder patients with psychotic symptoms and 120 ancestrally matched unscreened comparison subjects. Only patients with an age at onset below 35 years were included. Danish patients were interviewed using the semistructured Schedules for Clinical Assessment in Neuropsychiatry (version 2.1). Two psychiatrists made lifetime RDC diagnoses at the probable level of diagnosis. All bipolar patients met RDC and ICD-10 criteria for bipolar disorder. The 90-item OPCRIT checklist was also used for the patients from Denmark, and these patients also fulfilled Research Diagnostic Criteria and DSM-III-R criteria for bipolar disorder.
Genomic DNA was extracted from whole blood using standard methods. Fifteen microsatellite marker loci were initially selected from the Marshfield Centre for Medical Genetics database (http://research.marshfieldclinic.org/genetics) and from the Genome database (http://www.gdb.org). In addition, at the region where fine mapping was to be performed, a publicly available genomic sequence was examined for potential microsatellite repeat structures. These sequences were amplified in 32 samples, and the products were investigated for potential polymorphic markers. Seven novel microsatellites were identified using this approach. All genotyping was carried out by polymerase chain reaction (PCR) amplification of microsatellites using standard methods followed by electrophoresis. In both study centers, amplification was performed with one of the PCR primers being directly labeled with a fluorescent probe. In Denmark, the samples were then automatically loaded and electrophoresed using the POP 4 protocol for the ABI Prism 310 Genetic Analyser. GeneScan and GenoTyper programs were used to perform analyses of the allele sizes and allele scoring (Applied Biosystems, Foster City, Calif.). In the U.K. the amplified samples were loaded manually and electrophoresed on Li-Cor DNA4200 sequencers (Li-Cor Biosciences, Lincoln, Neb.). The GeneImagIR or SAGA-GT (Li-Cor) programs were used to perform analysis of the allele sizes and for allele scoring. In both centers, two researchers scored the genotypes independently and repeated any discrepant genotypes. Sequencing of exons and flanking intronic sequences was performed using the Li-Cor infrared fluorescent sequencing system. Sequencing products were arranged such that the guanosine dideoxy nucleotide terminated tracks from each of the samples were loaded together followed by the adenosine, thymidine, and cytidine. This method enables accurate visual scoring of potential mutations and allows recognition of polymorphisms in their heterozygote state. The validity of potential mutations was assessed by comparison with the gel image produced from simultaneous sequencing reactions primed from the opposite strand.
Some of the SNPs identified in the U.K. cohort were subsequently discovered to be identical to those logged in the public databases. All the SNPs identified so far have been genotyped in the U.K. cohort but not in the Danish cohort. SNPs rs4765108, 47783-ct, rs1194031, and 29818-insT were typed using the Pyrosequencing technology according to manufacturer’s instructions (Pyrosequencing, Sweden). SNPs rs2292446, rs7133178, and rs10773323 were typed using Taqman MGB probes according to manufacturer’s instructions (Applied Biosystems). SNPs Mette_1 and rs1212337 were typed by Kbiosciences (Kbiosciences, U.K.) using the Amplifluor SNP genotyping method (Serologicals Corp., Atlanta, Ga.), and the pufu insertion/deletion polymorphism (pufu in/del) was genotyped as for a microsatellite.
Allelic association between bipolar disorder and each marker was tested using the CLUMP program (18) . This performs a Monte Carlo evaluation of significance levels to produce an empirical p value for the observed chi-square statistics, since the asymptotic p value may be inaccurate for markers having large numbers of alleles. Four different chi-square statistics are calculated, labeled T1-T4: T1 is the standard Pearson chi-square statistic; T2 is the statistic obtained by collapsing together rare alleles; the T3 statistic is the maximum obtained by considering each allele separately against all the rest; the T4 statistic is obtained by grouping together all the alleles positively and negatively associated with disease. CLUMP analysis was performed on the two populations separately and then analysis of the Danish and U.K. cohorts combined was performed. Linkage disequilibrium between pairs of markers was determined using LDPAIRS, an accessory program available with the GENECOUNTING package. Pairs or trios of markers that were in linkage disequilibrium with one another were used for haplotype analysis. Haplotype analysis was carried out using the program GENECOUNTING, which uses an algorithm capable of analyzing both SNPs and microsatellites and has the advantage of being able to deal with missing data (19 , 20) . The program estimates haplotype frequencies from unphased, multilocus genotype data. Permutation testing was performed to assess the statistical significance of differences in estimated haplotype frequencies between patients and comparison subjects. Loci were studied in sets of two or three at a time. The genotype data were also analyzed separately by a 96-well DNA plate using SCANGROUP to test for differences in haplotype frequencies that may have occurred by systematic error in genotyping and thus may have led to type I error.
Results
Evidence for allelic association was first found in the cohort from Denmark, and this was used to test for association in the larger U.K. cohort. The following 31 novel and published microsatellite and SNP markers were genotyped: D12S1614, D12S342, AFMb332ZC1, D12S340, D12S1639, D12S324, D12S866, D12S386, 1634GT2, AFMb337ZD5, 1634tet, D12S1634, rs2292446, rs4765108, 307GT4, D12S307, rs7133178, pufu in/del, 47783-ct, Mette_1, rs1212337, rs10773323, rs1194031, 29818-insT, 307CA1, 307CA2, D12SDK2, D12SDK1, D12S1658, GATA41E12, and D12S2075. Primer sequences for the established microsatellite markers are available from the Genome database, and the sequence surrounding the published SNPs is available from the SNP database. We identified and genotyped the following new microsatellite and SNP markers: 1634GT2, 1634tet, 307GT4, pufu in/del, 47783-ct, Mette_1, rs10773323, 29818-insT, 307CA1, 307CA2, D12SDK2, and D12SDK1. The primer sequences for the new microsatellite markers or the sequence surrounding SNPs are shown in Table 1 as well as their positions on 12q24. All the SNPs and microsatellite markers were found to be in Hardy-Weinberg equilibrium in both the U.K. and Danish bipolar and comparison groups. The empirical levels of significance derived from the CLUMP analysis for microsatellite markers or allelic chi-square tests for SNPs are shown in Table 2 and graphically in Figure 1 . The location of these markers, the interval between markers, the number of alleles observed and the number of individuals genotyped are also shown in Table 2 . Four markers originally produced significant p values in the Danish sample: D12S1634, 307CA1, 307CA2, and D12S2075. Exactly the same region that was implicated in the Danish cohort was then tested in the U.K. cohort, and seven markers produced significant p values that did not need further correction for the presence of multiple alleles. These were 1634tet, 307GT4, D12S307, rs7133178, pufu in/del, 29818-insT, and D12SDK1 ( Table 2 ).

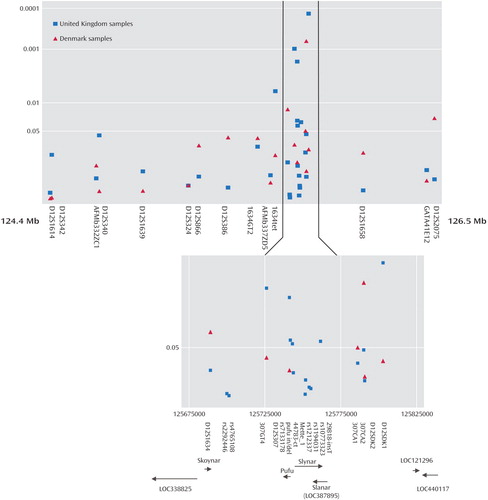
a The positions of the markers are shown according to their alignment with the May 2004 human reference sequence, which is based on NCBI Build 35. The most significant results obtained with each marker for the U.K. and Danish cohorts are shown. The candidate region is shown enlarged and includes the positions of the markers in base pairs from the tip of the short arm (p-tel), the most significant CLUMP empirical p value results from these markers, the position of predicted genes in the region, and the direction in which they are transcribed.
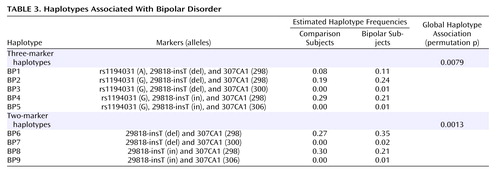
When allele counts for the Danish and U.K. cohorts were combined, six markers produced significant associations: D12S340 (p=0.042 [T3]), 1634GT2 (p=0.01 [T2]), 1634TET (p=0.004 [T2]), 307GT4 (p=0.021 [T3]), D12S307 (p=0.001 [T3]), and D12SDK1 (p=0.003 [T4]). Pairwise marker-to-marker linkage disequilibrium was determined using LDPAIRS. Absolute D′ and significance values for the linkage disequilibrium relationships are shown in Figure 2 . As expected, stronger linkage disequilibrium was observed in the region where marker density was greatest. Estimated haplotype frequencies for the bipolar and comparison groups using data from pairs or trios of markers that were in pairwise linkage disequilibrium were generated using GENECOUNTING. The most significant three-marker haplotype was found when combining data from markers rs1190431, 29818-insT, and 307CA1 (empirical p=0.0079). Individual two- and three-marker haplotypes, denoted BP1 through BP9, with estimated frequencies that were notably elevated or reduced in bipolar patients relative to comparison subjects are shown in Table 3 . Two-marker haplotype analysis using 29818-insT and 307CA1 produced a significant empirical p value of 0.0013 and estimated that 35% of bipolar patient chromosomes carry the 12 (BP6) haplotype and for this haplotype to occur in 27% of comparison chromosomes ( Table 3 ). This suggests that the BP6 haplotype may carry an elevated risk for bipolar disorder. The haplotype with alleles G, del, and 298 (BP2) as shown in Table 3 , generated from analysis of data from rs1194031, 29818-insT, and 307CA1 was estimated to occur in approximately 24% of bipolar patient chromosomes and in 19% of comparison chromosomes, again suggesting that this haplotype may carry an elevated risk for bipolar disorder. Similarly haplotype BP1 with A, del, and 298 was estimated to occur in 11% of bipolar patient chromosomes and 8% of comparison chromosomes. Additional haplotypes (BP3–5, BP7–9) that were estimated to be at altered frequency in bipolar patients relative to comparison subjects are also shown in Table 3 .

Discussion
The results presented here provide further support for the presence of a bipolar locus on 12q24. The region spanned by markers showing positive association covers 278 Kb and suggests that the mutation or mutations influencing susceptibility to bipolar disorder lie within this region. We obtained replication in the U.K. cohort with markers in exactly the same genomic region that was implicated in the Danish cohort. This is most evident with four markers in the U.K. cohort that were originally implicated in the Danish cohort. In the original Danish cohort all four markers gave significant empirical p values and in the U.K. cohort these markers were only significantly associated with nonempirical (asymptotic) T3 p values. Empirical p values from CLUMP tests were significant in both groups individually, with multiple markers in the same region, but these were not for the same markers in each of the two cohorts. Further correction for multiple loci in the single marker tests of association would be complicated by the fact that the markers showing association are in strong linkage disequilibrium with each other and represent nonindependent tests of association. The permutation tests of significance for haplotypic association do not need correction for multiple alleles or multiple loci.
We note that one marker, D12S2075, which was not in linkage disequilibrium with the other positive markers, also showed allelic association with bipolar affective disorder in a more distal position. This could be at a second chromosome 12 affective disorder susceptibility locus. The U.K. bipolar group has a power to detect allelic association of 0.99 at p<0.05 and of 0.91 at p<0.001 assuming complete linkage disequilibrium between a marker and disease allele when the minor marker allele frequency is less than 10% with a 5% difference in allele frequencies. If this allele frequency is as high as 50%, the power is 0.90 at p<0.05 with a 10% allele frequency difference but is only 0.41 at p<0.05 with a 5% allele frequency difference. The power of the Danish cohort is much less, and the results suggest that the main susceptibility alleles are widely distributed in European populations with it being well represented in the cohort from Denmark. The evidence from both the single marker analysis and haplotype tests of association indicates that the most strongly implicated region is surrounding the marker D12S307, which is placed within the AY070435 or Slynar gene.
There is the possibility that there is a separate bipolar susceptibility locus on 12q23-q24. In a French Canadian case/control study, microsatellite markers 5.6 and 4.5 Mb proximal to D12S307 were found to be associated with bipolar disorder (15) . These distances are not compatible with a single locus producing the evidence for association that we detected, because linkage disequilibrium with disease susceptibility alleles over such a large region in a complex disease would not occur. The marker D12S2075, 2 Mb distal to D12S307 (which we found to be associated with bipolar disorder), was also found to be associated in the French Canadian cohort (15) . This marker was also positively associated in the current Danish cohort, and this region also warrants further investigation. Another putative bipolar susceptibility locus 15 Mb proximal to D12S307 has been identified in a bipolar patient group from South Wales and the Midlands of England but is unreplicated as yet (16) . This study focused on a critical region identified in two pedigrees with bipolar disorder and Darier’s disease (11 , 16) , and an association with two microsatellites and six SNPs in a 150-Kb region was identified around the CUX2 and FLJ32356 locus on 12q23-q24. The evidence so far is consistent with there being three bipolar susceptibility loci in the 12q region.
The most promising candidate gene in the region implicated by us is denoted Slynar in the ACEVIEW database (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly [UniGene cluster Hs.369455]). ACEVIEW-generated gene names are the accepted names for genes that have not yet been well characterized. The region is not well characterized in terms of transcription, alternative splicing, and protein translation. So far there is one confirmed gene in the region and several other predicted genes that may not be independent of Slynar and which may be alternative transcripts ( Figure 1 ). A cDNA sequencing of Slynar transcripts has, so far, shown alternative splicing with six splice variants derived from fetal and adult human brain tissue, testis, pooled germ cell tumors, and kidney tumors. Slynar bears homology to a macaque brain-expressed gene, and its expression as an mRNA in the human brain has been confirmed by us (data to be published elsewhere). No known function has been attributed to this gene, although products of predicted translation products of some splice variants contain PSORT-predicted membrane localization motifs (21) .
Another candidate gene termed LOC387895 (UniGene cluster Hs.534660; Slanar in the ACEVIEW database) is antisense to Slynar and so may have a role in the control of Slynar expression (22) . Expression of LOC387895 in human brain tissue has also been confirmed by us (data to be published elsewhere), and 14 alternative splice variants have been isolated from various tissues. Pufu (ACEVIEW name; represented by a single EST [Genbank ID 27825752]) and LOC440117 (UniGene cluster Hs.127100) are two other predicted genes that are brain expressed. Pufu expression in the brain has also been confirmed by us, and LOC440117 has been isolated from a Medulla cDNA library. There are three other potential genes in the region: LOC338825 (UniGene cluster Hs.121392), LOC121296, and Skoynar. The sample in which this association has been detected has also been used to detect linkage disequilibrium with a new bipolar susceptibility locus at the TRPM2 and TSPEAR genes close to the telomere on the long arm of chromosome 21q22.3 (23) .
Both the chromosome 21q22.3 and 12q24 positive fine mapping results indicate that genetic loci implicated in family linkage studies are also increasing genetic susceptibility in bipolar patients selected without necessarily having a positive family history. Replication of the genetic association findings presented here in multiple samples is now needed to further confirm the fine mapping of this novel 12q24 bipolar susceptibility locus.
1. Blackwood DH, Visscher PM, Muir WJ: Genetic studies of bipolar affective disorder in large families. Br J Psychiatry Suppl 2001; 41:134–136Google Scholar
2. Craddock N, Jones I: Molecular genetics of bipolar disorder. Br J Psychiatry Suppl 2001; 41:S128–S133Google Scholar
3. Rifkin L, Gurling H: Genetic aspects of affective disorders, in Biological Aspects of Affective Disorders. Edited by Horton R, Katona C. London, Academic Press, 1991, pp 305–329Google Scholar
4. Dawson E, Parfitt E, Roberts Q, Daniels J, Lim L, Sham P, Nothen M, Propping P, Lanczik M, Maier W, Reuner U, Weissenbach J, Gill M, Powell J, McGuffin P, Owen M, Craddock N: Linkage studies of bipolar disorder in the region of the Darier’s disease gene on chromosome 12q23–24.1. Am J Med Genetics (Neuropsychiatric Genetics) 1995; 60:94–102Google Scholar
5. Ewald H, Degn B, Mors O, Kruse TA: Significant linkage between bipolar affective disorder and chromosome 12q24. Psychiatr Genet 1998; 8:131–140Google Scholar
6. Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G, Rollins DY, Moses T, Sanders AR, Karkera JD, Esterling LE, Zeng J, Ferraro TN, Guroff JJ, Kazuba D, Maxwell ME, Nurnberger JI, Gershon ES: A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. PNAS 1999; 96:5604–5609Google Scholar
7. Morissette J, Villeneuve A, Bordeleau L, Rochette D, Laberge C, Gagne B, Laprise C, Bouchard G, Plante M, Gobeil L, Shink E, Weissenbach J, Barden N: Genome-wide search for linkage of bipolar affective disorders in a very large pedigree derived from a homogeneous population in Quebec points to a locus of major effect on chromosome 12q23-q24. Am J Med Genet 1999; 88:567–587Google Scholar
8. Shink E, Morissette J, Sherrington R, Barden N: A genome-wide scan points to a susceptibility locus for bipolar disorder on chromosome 12. Mol Psychiatry 2005; 10:545–552Google Scholar
9. Curtis D, Kalsi G, Brynjolfsson J, McInnis M, O’Neill J, Smyth C, Moloney E, Murphy P, McQuillin A, Petursson H, Gurling H: Genome scan of pedigrees multiply affected with bipolar disorder provides further support for the presence of a susceptibility locus on chromosome 12q23-q24, and suggests the presence of additional loci on 1p and 1q. Psychiatr Genet 2003; 13:77–84Google Scholar
10. Degn B, Lundorf MD, Wang A, Vang M, Mors O, Kruse TA, Ewald H: Further evidence for a bipolar risk gene on chromosome 12q24 suggested by investigation of haplotype sharing and allelic association in patients from the Faroe Islands. Mol Psychiatry 2001; 6:450–455Google Scholar
11. Green E, Elvidge G, Jacobsen N, Glaser B, Jones I, O’Donovan MC, Kirov G, Owen MJ, Craddock N: Localization of bipolar susceptibility locus by molecular genetic analysis of the chromosome 12q23-q24 region in two pedigrees with bipolar disorder and Darier’s disease. Am J Psychiatry 2005; 162:35–42Google Scholar
12. Jacobsen NJ, Franks EK, Elvidge G, Jones I, McCandless F, O’Donovan MC, Owen MJ, Craddock N: Exclusion of the Darier’s disease gene, ATP2A2, as a common susceptibility gene for bipolar disorder. Mol Psychiatry 2001; 6:92–97Google Scholar
13. Ruiz-Perez VL, Carter SA, Healy E, Todd C, Rees JL, Steijlen PM, Carmichael AJ, Lewis HM, Hohl D, Itin P, Vahlquist A, Gobello T, Mazzanti C, Reggazini R, Nagy G, Munro CS, Strachan T: ATP2A2 mutations in Darier’s disease: variant cutaneous phenotypes are associated with missense mutations, but neuropsychiatric features are independent of mutation class. Hum Mol Genet 1999; 8:1621–1630Google Scholar
14. Abkevich V, Camp NJ, Hensel CH, Neff CD, Russell DL, Hughes DC, Plenk AM, Lowry MR, Richards RL, Carter C, Frech GC, Stone S, Rowe K, Chau CA, Cortado K, Hunt A, Luce K, O’Neil G, Poarch J, Potter J, Poulsen GH, Saxton H, Bernat-Sestak M, Thompson V, Gutin A, Skolnick MH, Shattuck D, Cannon-Albright L: Predisposition locus for major depression at chromosome 12q22–12q23.2. Am J Hum Genet 2003; 73:1271–1281Google Scholar
15. Shink E, Harvey M, Tremblay M, Gagne B, Belleau P, Raymond C, Labbe M, Dube MP, Lafreniere RG, Barden N: Analysis of microsatellite markers and single nucleotide polymorphisms in candidate genes for susceptibility to bipolar affective disorder in the chromosome 12Q24.31 region. Am J Med Genet B Neuropsychiatr Genet 2005; 5:50–58Google Scholar
16. Glaser B, Kirov G, Green E, Craddock N, Owen MJ: Linkage disequilibrium mapping of bipolar affective disorder at 12q23-q24 provides evidence for association at CUX2 and FLJ32356. Am J Med Genet B Neuropsychiatr Genet 2005; 132:38–45Google Scholar
17. McGuffin P, Farmer A, Harvey I: A polydiagnostic application of operational criteria in studies of psychotic illness: development and reliability of the OPCRIT system. Arch Gen Psychiatry 1991; 48:764–770Google Scholar
18. Sham P, Curtis D: Monte-Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet 1995; 59:97–105Google Scholar
19. Zhao JH, Lissarrague S, Essioux L, Sham PC: Genecounting: haplotype analysis with missing genotypes. Bioinformatics 2002; 18:1694–1695Google Scholar
20. Curtis D, Knight J, Sham PC: Program report: Genecounting support programs. Ann Hum Genet 2006; 70:277–279Google Scholar
21. Nakai K, Horton P: Psort: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 1999; 24:34–36Google Scholar
22. Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G: Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 2003; 21:379–386Google Scholar
23. McQuillin A, Bass NJ, Kalsi G, Lawrence J, Puri V, Choudhury K, Detera-Wadleigh SD, Curtis D, Gurling HMD: Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol Psychiatry 2006; 11:134–142Google Scholar

