
Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies
Abstract
Abstract
An emerging literature from epidemiologic, clinical, and preclinical investigations has provided evidence that gestational exposure to infection contributes to the etiology of schizophrenia. In recent years, these studies have moved from ecologic designs, which ascertain infection based on epidemics in populations, to investigations that have capitalized on reliable biomarkers in individual pregnancies. These studies have documented specific candidate infections that appear to be associated with an elevated risk of schizophrenia. Animal models of maternal immune activation inspired by this work have revealed intriguing findings indicating behavioral, neurochemical, and neurophysiologic abnormalities consistent with observations in schizophrenia. In parallel studies in humans and animals, investigators are working to uncover the cellular and molecular mechanisms by which in utero exposure to infection contributes to schizophrenia risk. In this review, the authors discuss and critically evaluate the epidemiologic literature on in utero exposure to infection and schizophrenia, summarize emerging animal models of maternal immune activation, and discuss putative unique and common mechanisms by which in utero exposure to infection alters neurodevelopment, potentially increasing susceptibility to schizophrenia. The promise of this work for facilitating the identification of susceptibility loci in genetic studies of schizophrenia is illustrated by examples of interaction between in utero exposure to infection and genetic variants. The authors then elaborate on possible implications of this work, including the use of preventive measures for reducing the incidence of schizophrenia. Finally, they discuss new approaches aimed at addressing current challenges in this area of research.
For nearly 100 years, infectious microbes dominated discourse on the causes of disease. Fueled by the biomedical revolution and consequent advances in several other disciplines of medical research, however, interest in microbial pathogens beyond their classical role in infectious diseases began gradually to wane. Rather, medical research on diseases other than those considered to be infectious in nature continued to favor genetic and nonmicrobial etiologies of human illness.
Yet, in recent years, a proliferation of studies on infection as a risk factor for schizophrenia has emerged. If schizophrenia is a neurodevelopmental disorder, a hypothesis that is supported by converging evidence from several disciplines of research, then infection is a likely candidate risk factor, given that microbial pathogens have long been known to cause congenital brain anomalies. Rubella, herpes simplex virus, cytomegalovirus, toxoplasmosis, and other infections are potent disrupters of fetal neurodevelopment leading to abnormalities of brain and behavior, including mental retardation, learning disabilities, and hypoplasia of several brain regions (1). Consequently, investigators began to examine whether in utero exposure to infection was related to risk of schizophrenia. In the following two sections, we review the major ecologic and birth cohort studies of prenatal exposure to infection and schizophrenia. These studies were identified through searches of the PubMed database; the literature review was conducted from November 2008 to March 2009. Search terms included combinations of the following: prenatal, in utero, prenatal or in utero infection, prenatal or in utero influenza, prenatal or in utero exposure, schizophrenia, psychotic disorders, psychosis, neurodevelopment, infection, influenza, etiology, epidemic, pandemic, cohort, birth cohort, epidemiology, and reproductive outcomes. The search covered titles and abstracts and was restricted to English-language publications. References from studies retrieved were reviewed to identify additional articles. Studies were not limited to any particular research design.
Epidemiologic Evidence: Studies Based on Ecologic Data
The earliest studies of prenatal infection and schizophrenia were derived from studies that used ecologic data, namely, influenza epidemics in populations, to define exposure status (2) (Table 1). Initial studies provided evidence consistent with an association between second-trimester exposure to influenza epidemics and schizophrenia; further investigations, however, some of which were larger and featured more complete case ascertainment, failed to replicate the association. While similarly designed studies of other infectious agents have suggested potential relationships with schizophrenia, the effects have been generally weak, and few replication attempts have been made (2) (Table 2).
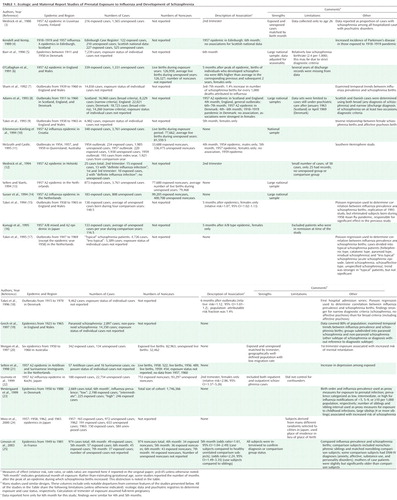 |
 |
These results led many to question whether prenatal exposure to influenza and other infections is a risk factor for schizophrenia. Unfortunately, inadequate consideration was given to significant limitations of this work, the most prominent of which was diagnostic misclassification of influenza. In nearly all studies of prenatal influenza and schizophrenia, the presence of the exposure was based solely on whether an individual was in gestation at the time of an influenza epidemic, with no confirmation of maternal influenza infection during pregnancy. Hence, discrepant findings between studies may have resulted from nondifferential misclassification of influenza exposure, which biases effect sizes toward the null. For example, by relying solely on dates of birth to define exposure to influenza, approximately 70% of individuals who were in gestation during the 1957 type A2 influenza epidemic but were unexposed would have been misclassified as having been exposed (31).
Epidemiologic Evidence: Birth Cohort Studies
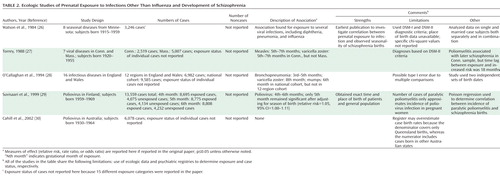
Consequently, a new approach was necessary to substantiate a link between prenatal exposure to infection and schizophrenia. Rather than relying on reports of epidemics in populations, we and other groups sought to document infection by measuring biomarkers in individual pregnancies and to relate confirmed exposure to the development of schizophrenia among individuals followed up into the age of risk for the disorder. This objective was achieved by capitalizing on well-characterized birth cohorts in which archived biological specimens or clinical diagnoses of infection were obtained during pregnancy and early life, and schizophrenia among offspring was systematically diagnosed following longitudinal assessment of offspring. We focus below on the major serological studies of prenatal infection and schizophrenia; further details are provided in Table 3. Studies based on infections identified from prospective maternal examinations and obstetric records are summarized in Table 4.
 |
 |
Influenza
Serologically documented prenatal exposure to influenza was examined in relation to schizophrenia in the Prenatal Determinants of Schizophrenia study (48), which was based on the birth cohort of the Child Health and Development Study (CHDS). This population-based cohort was born from 1959 to 1967 in Alameda County, Calif., and followed up for schizophrenia and other schizophrenia spectrum disorders in adulthood (Table 3). This investigation had several advantages, including archived maternal serum specimens prospectively drawn during pregnancy and stored frozen in a central repository; mothers belonging to the same health plan, which facilitated identification and follow-up of offspring with schizophrenia; and diagnoses confirmed with structured research interviews and psychiatric record review. Additional methodologic strengths included continuous follow-up assessments of offspring for schizophrenia, which allowed for adjustment for nondifferential loss to follow-up, and comparison subjects who were representative of the source population from which the cases were derived, which diminished the potential for bias due to loss to follow-up.
In a nested case-control study based on this cohort (37), our group demonstrated a threefold elevation in risk of schizophrenia following influenza exposure during the first half of gestation. For first-trimester exposure, the risk of schizophrenia was increased sevenfold. No elevated risk of schizophrenia was observed for influenza exposure during the second half of gestation, which suggests that these effects were specific to the period from early to midgestation.
Toxoplasma gondii (T. gondii)
T. gondii is a ubiquitous intracellular parasite that has been associated with several congenital CNS anomalies and more subtle delayed neurologic sequelae (1, 49). In the CHDS cohort, the risk of schizophrenia among individuals who were exposed in utero to elevated maternal T. gondii immunoglobin G (IgG) antibody, based on assay of archived maternal sera, was more than twice that of comparison subjects (38) (Table 3). A similar finding was demonstrated in an independent sample from Denmark in which T. gondii IgG was assayed in filter paper blood spots collected from offspring with schizophrenia and comparison offspring within 1 week of birth (45). Since T. gondii IgG antibody in infant blood almost certainly derived from the maternal rather than the fetal immune response, the latter finding can be considered a replication.
Herpes Simplex Virus Type 2 (HSV-2)
HSV-2 is a sexually transmitted virus, and maternal-offspring transmission generally occurs during passage through the birth canal. Neonatal exposure to HSV-2 is associated with congenital anomalies similar to those of gestational exposure to T. gondii, including neuropsychiatric outcomes (50). Three studies (32, 34, 40) have specifically investigated the relationship between prenatal exposure to HSV-2 and risk of schizophrenia among offspring. Two of these studies were derived from selected sites of the Collaborative Perinatal Project (CPP), a multisite study of population-based birth cohorts born from 1959 to 1967 throughout the United States that featured a number of methodologic advantages over earlier studies. Similar to the CHDS, archived maternal samples were prospectively drawn throughout pregnancy and stored in a central repository. The CPP also included follow-up for psychotic disorders in offspring at psychiatric treatment facilities using specified protocols and confirmation of diagnoses by psychiatric interviews and medical record reviews.
In the first of these studies (32), in the Providence, R.I., cohort of the CPP, maternal IgG antibody levels to HSV-2 were associated with a significantly elevated risk of psychosis (both nonaffective and affective) in offspring. Offspring in the highest quartile and decile for maternal HSV-2 antibody levels had odds ratios of 3.4 and 4.4, respectively, for risk of schizophrenia. In a much larger follow-up study (34), which included 200 case subjects with psychotic disorders and more than 500 matched comparison subjects from three cohorts of the CPP (Boston, Providence, and Philadelphia), a statistically significant 1.6-fold elevation in risk of psychosis and a significant 1.8-fold elevation in risk of schizophrenic psychoses were observed among offspring of mothers who were seropositive for HSV-2. The elevated risk was confined to offspring of seropositive mothers who did not regularly use contraception and had frequent intercourse.
In an investigation of prenatal HSV-2 exposure and schizophrenia based on the CHDS birth cohort that included 60 case subjects and 120 comparison subjects, these associations were not replicated (40), with odds ratios near 1 for all analyses. The discrepant results between the CPP study and the CHDS may be accounted for by several methodologic differences. First, the CPP cohort included a larger proportion of African Americans than the CHDS cohort. As noted by Buka et al. (34), the higher prevalence of HSV-2 in this ethnic group may have provided greater statistical power to detect an association in the overall sample. The number of cases of schizophrenic psychoses in the latter CPP study (N=108) was also significantly greater than that of the CHDS cohort (N=60), further increasing statistical power. Hence, the null results from the CHDS may have been due in part to lack of power to detect an association between prenatal HSV-2 exposure and schizophrenia.
The second difference is related to exposure status. In the two CPP studies, the definition of exposure in the primary analyses was based on differing criteria: the first study restricted the measure of exposure to maternal HSV-2 IgG antibody levels, while the reported data of the latter study were based on the presence of maternal seropositivity to HSV-2 (although it was noted that quantitative data from that study were consistent with the results based on seropositivity). In the study from the CHDS cohort, no associations were demonstrated regardless of whether the exposure was defined in terms of HSV-2 seropositivity or IgG antibody levels.
Third, loss to follow-up could not be quantified in the CPP because of lack of access to population denominators over the follow-up period. While significant loss to follow-up also occurred in the CHDS, bias was probably more likely to have been mitigated in that cohort by the use of survival analytic methods, which was made possible by continuous follow-up of the cohort.
Finally, the CHDS, unlike the CPP, did not have access to data on sexual risk behaviors. Conceivably, associations between HSV-2 and schizophrenia may have been demonstrated if this more vulnerable subgroup had been analyzed separately, as in the CPP study.
Cytokines
Cytokines are a family of soluble polypeptides that play an essential role in the immune response as the systemic mediators of the host response to infection. Hence, these molecules represent robust markers of infectious and inflammatory conditions (51). In an investigation based on the CHDS birth cohort (36), we observed a nearly twofold elevation in maternal levels of interleukin-8 (IL-8), a proinflammatory chemokine (chemokines are a subclass of the cytokine superfamily), during the second trimester and the early third trimester among pregnancies giving rise to offspring with schizophrenia, relative to matched comparison subjects (Table 3). In the sample from the Providence site of the CPP cohort, levels of tumor necrosis factor-α (TNF-α), a proinflammatory cytokine, at the time of birth were significantly increased among mothers of offspring with psychosis (nonaffective and affective psychoses combined) relative to comparison subjects (33).
Although the reasons for the discrepant findings between the two studies are not entirely clear, some potential explanations may be considered. First, in the CPP study, the mean maternal IL-8 level was not significantly elevated in pregnancies that gave rise to offspring with psychoses, but analyses that classified IL-8 as a categorical variable revealed a "significant and graded association" between maternal serum IL-8 levels and offspring adult psychosis (although the data corresponding to the latter finding were not reported) (32). With regard to other cytokines, the two studies were in agreement with regard to maternal interleukin-1β (IL-1β) and interleukin-6 (IL-6), neither of which was associated with schizophrenia or psychotic disorders, and maternal interleukin-2 (IL-2), which was examined only in the CPP study, was not associated with psychoses.
Causal Mechanisms: Unique Effects of Infections
Both epidemiologic and preclinical studies promise to shed light on causal mechanisms by which prenatal exposure to infection might lead to schizophrenia. One school of thought contends that individual infections act to influence risk of schizophrenia by unique effects. This hypothesis is supported by the fact that infections associated with schizophrenia differ significantly from one another in several respects, including duration, antigenicity and antibody response, capacity to traverse the placenta, gestational specificity on known congenital outcomes, and effects on brain development. The alternative hypothesis, reviewed in the next section, is that infections act via one or more common mechanisms to increase vulnerability to schizophrenia.
While a detailed review of the individual characteristics and pathogenic effects of each infection is beyond the scope of this article, we discuss, as one example, potential mechanisms by which toxoplasmosis might act to increase schizophrenia risk. Mechanisms relevant to active primary infection by T. gondii appear to be unlikely, given that maternal or neonatal seropositivity to T. gondii IgM-specific antibody, a robust indicator of recently acquired infection, was not observed in two studies of T. gondii and schizophrenia (38, 45). A second potential mechanism involves reactivation of a previous infection, resulting in an inflammatory response in the developing fetal brain. An increase in maternal IgG antibody can result from this anamnestic response. A third possible explanation for the finding is a dormant T. gondii infection, which may be accompanied by elevated T. gondii IgG antibody up to many years after the infection. Neuropathologic effects could potentially ensue from exposure to this antibody, which can cross the placenta. Intriguingly, elevated total IgG antibody has teratogenic effects in certain autoimmune disorders (52).
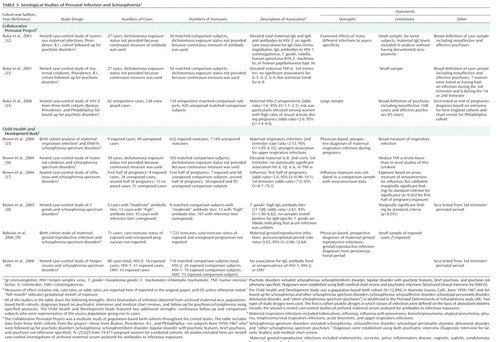
Preclinical Studies of Prenatal Exposure to Influenza
Preclinical approaches to infection with a specific microbe have thus far been restricted to inoculation of pregnant rodents with influenza, followed by behavioral, neurophysiologic, molecular, and neuroanatomic assessments (53, 54). In offspring from pregnancies in which intranasal infusion of influenza in midpregnancy was administered, decreased exploratory behavior, diminished contact with novel objects, reduced "social" behavior, and prepulse inhibition deficits to acoustic startle were observed (54). The prepulse inhibition deficit was reversed following treatment with clozapine and chlorpromazine. Neonatal exposure to influenza has been associated with significant reductions in reelin-positive Cajal-Retzius cells in several cortical layers and the hippocampus but without changes in calretinin and neuronal nitric oxide synthase, which suggests that the effect is not due solely to greater cell death but also to abnormal reelin production (53). Decreased cortical and hippocampal area and an increase in pyramidal cell density in cortex were also observed in neonatally infected mice.
Causal Mechanisms: Common Effects of Infection
In an alternative, more parsimonious model, infections are postulated to act through common pathways to alter fetal brain development and increase vulnerability to schizophrenia. Gilmore and Jarskog (55) proposed that one such common mechanism involves infection-induced cytokines, and this hypothesis has now been extensively tested in animal models based on the seminal findings of Patterson (56) and the work of other groups.
Clinical Evidence
While physiologic levels of cytokines play critical roles in normal brain development, elevations of these inflammatory mediators secondary to maternal infection increase vulnerability to developmental brain damage (57). Intrauterine exposure to elevated levels of infection-induced cytokines has been associated with chorioamnionitis, periventricular leukomalacia, and cerebral palsy. These abnormalities may develop as a result of several potential mechanisms, including stimulation of microglia and astroglia to produce nitric oxide and excitatory amino acids and disrupted maturation of oligodendrocytes, potentially contributing to white matter abnormalities, which have been associated with schizophrenia (58).
Most infections are associated with increases in several cytokines, including IL-8 and TNF-α, both of which have been associated with schizophrenia and other psychotic disorders, as reviewed above. Maternal serum IL-8 levels are associated with histologic chorioamnionitis in term infants, and maternal and fetal levels of IL-8 have been significantly correlated with one another. IL-8 belongs to the chemokine family and appears to be especially important for neutrophil attraction as well as discharge of lysosomal enzymes from neutrophils, leading to oxygen free radicals. TNF-α has also been associated with chorioamnionitis (59), and increased amniotic fluid levels of this cytokine have been associated with fetal infection (60). Polymorphisms in TNF-α genes have also been associated with schizophrenia (61).
Preclinical Evidence: Studies of Maternal Immune Activation
The vast majority of preclinical studies of infection have implemented an experimental paradigm involving maternal immune activation, as reviewed by Patterson (56).
Two agents have been used to induce immune activation during pregnancy: polyinosinic:polycytidylic acid (poly I:C), a synthetic analogue of double-stranded RNA, which mimics a viral infection, and lipopolysaccharide, a bacterial cell wall endotoxin. Interestingly, several brain and behavioral deficits in offspring were similar regardless of whether they resulted from maternal influenza or poly I:C (54). Offspring exposed to maternal immune activation also displayed dopaminergic abnormalities, including elevated amphetamine-induced locomotion, increased striatal dopamine release, and disrupted latent inhibition, the last of which was reversed by clozapine, suggesting elevated sensitivity of subcortical and mesolimbic dopaminergic pathways. Maternal immune activation exposure also resulted in increased locomotion after administration of the N-methyl-D-aspartic acid antagonist MK-801, which is consistent with glutamatergic models of schizophrenia (62, 63). In several preclinical maternal immune activation models, the observed phenotypes were not demonstrated during the juvenile period but emerged when testing occurred during adulthood, possibly modeling the developmental time course of the onset of schizophrenia.
Accumulating evidence from preclinical studies suggests potential mechanisms by which elevated cytokines might contribute to the observed brain and behavioral abnormalities implicated in schizophrenia. For example, direct treatment of embryonic rat cortical cultures with IL-1β and TNF-α led to dose-dependent decreases in immunoreactivity for MAP-2 antibody (64), which suggests that cytokines decrease cerebral cortical neuronal survival during brain development.
Although both maternal and fetal cytokines are elevated after immune activation, few studies have tested for a direct causal connection between cytokines and brain and behavioral abnormalities. Recently, Smith et al. (65) provided intriguing evidence supporting an important role for IL-6 in these outcomes in an animal model. They found that a single maternal injection of IL-6 in midpregnancy resulted in prepulse inhibition and latent inhibition deficits in adult offspring. Most of these effects were prevented after administration of anti-IL-6 antibody and by an IL-6 genetic knockout. While these findings do not prove that IL-6 is the sole contributor to the schizophrenia-related phenotypes resulting from maternal immune activation, they suggest a key role for this cytokine in the pathogenic process.
Gene-Environment Interplay
It has been widely acknowledged that genes and environmental exposures probably act in tandem to increase the risk of neuropsychiatric disorders, including schizophrenia. A gene-infection interaction in schizophrenia would be tested by assessing whether the presence of a susceptibility gene increases the effect of a prenatal infectious exposure on the risk of this disorder. Although no previous study has tested for interactions between individual susceptibility genes and prenatal infections in schizophrenia, a recent investigation using data from a large Finnish cohort revealed that prenatal exposure to pyelonephritis was demonstrated to have a fivefold greater effect on risk of schizophrenia among individuals with a family history of psychosis compared to those with no family history (47).
Genetic Effects on Neurodevelopment
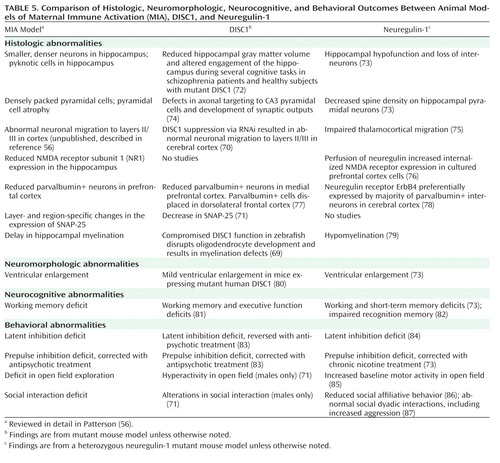
As reviewed in detail elsewhere, a considerable number of putative vulnerability genes for schizophrenia have been identified, and several of these associations have been replicated in multiple studies (66, 67). While many of these genes will not be directly involved in infectious or inflammatory processes, they may act in concert with prenatal infection to increase liability to schizophrenia by disrupting neurodevelopmental events. Intriguingly, evidence is emerging that two prominent putative susceptibility genes—DISC1 and neuregulin—appear to play critical roles in brain development (68–71). Polymorphisms of these two genes have been associated with schizophrenia in several populations throughout the world, and mutations in these genes have resulted in brain and behavioral anomalies similar to those observed in the maternal immune activation model.
Table 5 presents comparisons of specific brain and behavioral anomalies observed in studies of animal models of maternal immune activation, DISC1, and neuregulin. Animal model studies of DISC1 and neuregulin-1 were identified through searches of the PubMed database conducted from November 2008 to May 2009. Search terms were derived from the characterization of maternal immune activation as described in a recent comprehensive review by Patterson (56). To limit the scope of the material, only DISC1 and neuregulin-1 studies that described phenotypes that were similar or related to the maternal immune activation model as described in Patterson (56) were included in Table 5. Additional search strategies were employed as described at the start of this article.
 |
Genetic Effects on the Immune Response
In a second example of gene-environment interaction, infection alters the effect of genes that are central to the immune response. A group of particularly strong candidate genes that may be involved in this process are those that encode major histocompatibility complex (MHC) class I proteins, which present antigens to T lymphocytes at the cell surface. These molecules are enriched at the synapse, are required for normal synaptic function and synaptic remodeling, and play a particularly important role in graded fine-tuning of plasticity; their expression is regulated by cytokines (88). Under this model, prenatal infection and the consequent cytokine response may act to modify MHC class I function to a greater degree among individuals who are genetically predisposed to decreased function of this class of molecules, resulting in reduced synaptic plasticity, which has been postulated to play an important role in the pathophysiology of schizophrenia (89). Interestingly, genetic variants in the extended MHC represented one of the few results to meet genome-wide significance in three recent large-scale genome-wide association studies (90–92).
Attributable Proportion and Gene Identification
A concept that is particularly salient to the interpretation of gene-environment interaction is that of attributable proportion, which is defined as the proportion of case subjects in the population who would not have developed the disease if a cause were removed (93). This is theoretically similar to the "causal pie" models elaborated by other authors (93, 94). One key implication of this notion is that in the presence of gene-environment interaction, it would be an oversimplification to conclude that individual environmental risk factors or susceptibility genes "explain" a particular proportion of cases in the population.
This concept has implications for facilitating gene identification. While recent molecular genetic studies promise to interrogate the genome with greater precision than has ever been realized, it appears doubtful, in our view, that a sole focus on gene-finding will lead to definitive causes if gene-environment interaction plays a significant role. If the effect of certain susceptibility genes on risk of schizophrenia is dependent on prenatal infection, then an alternative and potentially more productive approach to gene identification would be to scan the genome in a subset of the population that has been exposed to the infection in question.
Implications of Research on Prenatal Exposure to Infection and Schizophrenia
Implications for Prevention
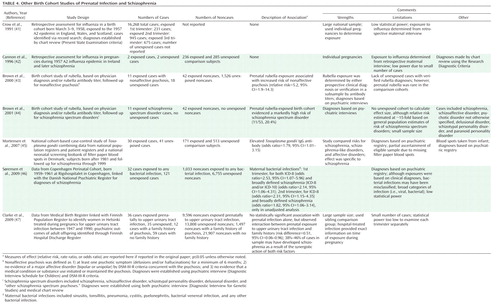
Investigations of prenatal exposure to infection in schizophrenia may have implications for prevention of the disorder. In this regard, the concept of attributable proportion also offers the potential to estimate the relative importance of preventive efforts. Attributable proportion is dependent on the magnitude of the effect of a component cause, the prevalence of the component cause in the population studied, and the nature of the interaction between component causes (94). Given that influenza, T. gondii, and genital/reproductive infections are highly prevalent and associations between prenatal exposure to each of these pathogens and schizophrenia were observed in our previous studies, we calculated the attributable proportion corresponding to the individual and combined effects of these infections using the data acquired from these studies and the standard published formula (94). The results are presented in Table 6. Given that little overlap was observed between these infections in the schizophrenia case and comparison subjects of the CHDS cohort (A.S. Brown et al., unpublished data), the attributable proportion for the combined effects of these infections is approximately the sum of the individual values. Hence, based on our data, we estimate that nearly one-third of schizophrenia cases could have been prevented if these infections had been entirely eliminated from the pregnant population. While it is unlikely that complete eradication of any infectious pathogen can be practically achieved, and although this finding clearly requires replication in an independent sample, this result raises the possibility that reducing the occurrence of prenatal exposure to influenza infection could nonetheless bring about an appreciable decline in the prevalence of schizophrenia.
 |
Although estimates of attributable proportion have not been calculated for most putative risk factors for schizophrenia, the potential contribution of infection appears to be comparable to many of these other factors, based on similarities in effect sizes and population prevalence. For example, with regard to paternal age greater than 40, most groups have demonstrated effect sizes of at least twofold, and Malaspina et al. (95) calculated an attributable proportion of 26.6%. The effect sizes for low birth weight/prematurity and hypoxia, obstetric exposures that have been well replicated in studies of schizophrenia, are generally similar in magnitude to those demonstrated in studies of prenatal exposure to infection (96).
If gene-environment interaction contributes to the attributable proportion estimate for schizophrenia, as discussed in the preceding section, then "correcting" the genetic defects may not be necessary in order to eliminate subsets of schizophrenia cases that are accounted for by interactions between genetic and environmental factors. Rather, elimination of the environmental factor, such as prenatal infection, may be a more practical and feasible approach in many cases than one that targets the gene or its translated protein.
Recommendations for Public Health Practice
If associations between prenatal exposure to infection and schizophrenia are independently replicated in other cohorts, there may be cause for optimism given that many microbial infections are either treatable or preventable. Indeed, it is worth considering that the overall decline in bacterial illnesses and initiation of immunization programs may have reduced the incidence of schizophrenia since the 1950s, as suggested by Suvisaari et al. (97).
With regard to treatment, many genital/reproductive system microbes are readily eliminated by antibiotics. Hence, increased surveillance and treatment of the reproductive-age population for these infections may have the potential to reduce the risk of schizophrenia. This class of infections includes sexually transmitted diseases, such as HSV-2, which are preventable by barrier contraceptives.
The prevention of influenza has been a preoccupation of modern medicine for the past several decades. The elevated vulnerability of the mother and fetus to influenza has resulted in the inclusion of pregnant women as a group to be targeted for the vaccination. In our view, this strategy has potential benefits but possibly also certain risks. Routine administration of influenza vaccine during pregnancy will clearly mitigate the potential for exposure to influenza. Nonetheless, this benefit will need to be weighed against the expected exposure in the population of large numbers of pregnant women, most of whom would never have developed influenza, to an agent that may induce a cytokine response. This could have the undesired effect of inducing unnecessary damage to the developing fetal brain. These questions are particularly salient in light of the recent emergence of the H1N1 flu epidemic.
It has long been known that toxoplasmosis during pregnancy is preventable by relatively straightforward hygienic approaches, including avoidance of contamination from cat litter boxes and gardening and thorough cooking of meats, poultry, and fish to kill T. gondii oocysts. It is worth bearing in mind, however, that schizophrenia was shown to be related to elevated maternal T. gondii IgG levels (38, 45), not the presence of the parasite itself, which suggests that most infections were acquired before pregnancy. Since acquisition of T. gondii can occur throughout the life course, it may be worth considering measures to reduce exposure to this infection in the general population as early as childhood as well as to investigate measures of screening for, and reducing levels of, T. gondii IgG in women of reproductive age, especially those who are planning a pregnancy.
A New Proposed Research Agenda
Infectious Exposures and Molecular Genetic Studies
In our view, several strategies for research will be critical for advancing and refining the hypothesis that prenatal exposure to infection plays a role in the etiology of schizophrenia. First, we recommend the incorporation of prospectively documented, specific, and validly measured infectious exposures into molecular genetic studies of gene-environment interaction. While relatively few in number, approaches to identifying gene-environment interaction that have modeled genes in the same way as environmental exposures have yielded potentially groundbreaking findings (98, 99). Genome-wide association studies have generally yielded small effect sizes of individual genetic variants (100, 101). If the findings of existing gene-environment interaction studies represent a valid gauge of what we might expect from studies of susceptibility genes and prenatal infections, we anticipate that enriching the sample for the presence of these exposures will enhance the "signal" of susceptibility genes, particularly those that influence brain development or are involved in pathogenic mechanisms related to microbial etiologies. Other promising approaches to characterizing genetic influences, including the identification of copy number variants and epigenetic effects, should also be facilitated by research designs that document specific prenatal infections that have been demonstrated to interact with the genome and epigenome.
Translational Approaches
The adoption of translational approaches that capitalize on findings from epidemiologic research has considerable potential for integrating etiologic and pathophysiologic research in schizophrenia. As reviewed above, animal models have yielded evidence that prenatal exposure to infection causes phenotypes that appear to resemble brain and behavioral anomalies observed in schizophrenia. In our view, translational research aimed at leveraging this line of inquiry toward elucidating causal mechanisms by which neurodevelopment is altered by these prenatal insults should be given high priority. This will require interdisciplinary efforts between epidemiologists, geneticists, and molecular/cellular neuroscientists.
Windows of Vulnerability
In our opinion, greater attention needs to be devoted to understanding critical windows of vulnerability to infectious insults and the effects of these exposures on developmental trajectories. As exemplified by the epidemiologic studies reviewed above, the gestational timing of infection might be a critical factor influencing the risk of schizophrenia. Unfortunately, there has been a persistence of the notion that the effect of prenatal infection is isolated to midgestation. We contend that a more complicated scenario, in which the developmental window of vulnerability varies by type of infectious exposure, is more consistent with the empirical evidence and with our understanding of neurodevelopmental influences in general.
Conclusions
Epidemiologic studies have yielded evidence suggesting that prenatal exposure to infection plays a role in the etiology of schizophrenia. The use of biomarkers of prenatal infection in rigorous epidemiologic designs of birth cohorts followed into the age of risk for schizophrenia is a potentially useful new approach to either confirming or refuting findings from ecologic studies, identifying new microbial risk factors, and uncovering pathogenic mechanisms. In our view, research efforts aimed at discovering interactions between prenatal exposure to infection and susceptibility genes hold promise toward improving our understanding of both the genetic and the environmental bases of schizophrenia. The development of effective treatments for infectious diseases was one of the greatest successes of 20th-century medical science and was a major factor in the advent of modern medicine. The identification of microbial risk factors required rigorous and painstaking epidemiologic studies, and we expect that efforts in the 21st century to discover infectious causes of schizophrenia, as well as other diseases previously considered to be noninfectious, will be no less challenging. Nonetheless, given the high prevalence of microbial disease in pregnancy and ready access to existing treatments and preventive approaches to combat infectious illness, it is conceivable that these efforts will ultimately have an appreciable impact on the incidence and burden of schizophrenia in the global population.
1 Remington JSKlein JOWilson CBBaker CJ (eds): Infectious Diseases of the Fetus and Newborn Infant, 6th ed. Philadelphia, Elsevier Saunders, 2006 Google Scholar
2 : Prenatal infections, in Prenatal Exposures: Psychological and Educational Consequences for Children. New York, Springer, 2008, pp 57–86 Google Scholar
3 : Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry 1988; 45:189–192 Crossref, Medline, Google Scholar
4 : Maternal influenza in the etiology of schizophrenia. Arch Gen Psychiatry 1989; 46:878–882 Crossref, Medline, Google Scholar
5 : Exposure to influenza epidemics during gestation and adult schizophrenia: a 40-year study. Arch Gen Psychiatry 1990; 47:869–874 Crossref, Medline, Google Scholar
6 : Season of birth in schizophrenia: evidence for confinement of an excess of winter births to patients without a family history of mental disorder. Br J Psychiatry 1991; 158:764–769 Crossref, Medline, Google Scholar
7 : Schizophrenia following pre-natal exposure to influenza epidemics between 1939 and 1960. Br J Psychiatry 1992; 160:461–466 Crossref, Medline, Google Scholar
8 : ERP abnormalities during semantic processing in schizophrenia. Schizophr Res 1993; 10:247–257 Crossref, Medline, Google Scholar
9 : Does prenatal influenza divert susceptible females from later affective psychosis to schizophrenia? Acta Psychiatr Scand 1993; 88:328–336 Crossref, Medline, Google Scholar
10 : Schizophrenia and prenatal exposure to the 1957 A2 influenza epidemic in Croatia. Am J Psychiatry 1994; 151:1496–1498 Link, Google Scholar
11 : Does influenza cause schizophrenia? a five year review. Aust NZ J Psychiatry 1995; 29:23–31 Crossref, Medline, Google Scholar
12 : Prenatal influenza infections and adult schizophrenia. Schizophr Bull 1994; 20:263–267 Crossref, Medline, Google Scholar
13 : Evidence against maternal influenza as a risk factor for schizophrenia. Br J Psychiatry 1994; 164:674–676 Crossref, Medline, Google Scholar
14 : No relation between risk of schizophrenia and prenatal exposure to influenza in Holland. Am J Psychiatry 1994; 151:922–924 Link, Google Scholar
15 : Prenatal exposure to influenza and the development of schizophrenia: is the effect confined to females? Am J Psychiatry 1994; 151:117–119 Link, Google Scholar
16 : Schizophrenia following in utero exposure to the 1957 influenza epidemics in Japan. Am J Psychiatry 1995; 152:450–452 Link, Google Scholar
17 : Schizophrenia risk in women from in utero exposure to influenza (letter). Am J Psychiatry 1995; 152:150–151 Link, Google Scholar
18 : Relationship between in utero exposure to influenza epidemics and risk of schizophrenia in Denmark. Biol Psychiatry 1996; 40:817–824 Crossref, Medline, Google Scholar
19 : Maternal exposure to influenza and paranoid schizophrenia. Schizophr Res 1997; 26:121–125 Crossref, Medline, Google Scholar
20 : Influenza epidemics and incidence of schizophrenia, affective disorders, and mental retardation in Western Australia: no evidence of a major effect. Schizophr Res 1997; 26:25–39 Crossref, Medline, Google Scholar
21 : Prenatal exposure to influenza and schizophrenia in Surinamese and Dutch Antillean immigrants to the Netherlands. Schizophr Res 1998; 30:101–103 Crossref, Medline, Google Scholar
22 : Schizophrenia and the influenza epidemics of 1957 in Japan. Biol Psychiatry 1999; 46:119–124 Crossref, Medline, Google Scholar
23 : Exposure to prenatal and childhood infections and the risk of schizophrenia: suggestions from a study of sibship characteristics and influenza prevalence. Arch Gen Psychiatry 1999; 56:993–998 Crossref, Medline, Google Scholar
24 : No relationship between schizophrenic birth and influenza epidemics in Japan. J Psychiatr Res 2000; 34:133–138 Crossref, Medline, Google Scholar
25 : Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003; 107:331–335 Crossref, Medline, Google Scholar
26 : Schizophrenic birth seasonality in relation to the incidence of infectious diseases and temperature extremes. Arch Gen Psychiatry 1984; 41:85–90 Crossref, Medline, Google Scholar
27 : Stalking the schizovirus. Schizophr Bull 1988; 14:223–229 Crossref, Medline, Google Scholar
28 : The relationship of schizophrenic births to 16 infectious diseases. Br J Psychiatry 1994; 165:353–356 Crossref, Medline, Google Scholar
29 : Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am J Psychiatry 1999; 156:1100–1102 Abstract, Google Scholar
30 : No significant association between prenatal exposure poliovirus epidemics and psychosis. Aust NZ J Psychiatry 2002; 36:373–375 Crossref, Medline, Google Scholar
31 : Influenza. New York, Plenum, 1987 Crossref, Google Scholar
32 : Maternal infections and subsequent psychosis among offspring. Arch Gen Psychiatry 2001; 58:1032–1037 Crossref, Medline, Google Scholar
33 : Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun 2001; 15:411–420 Crossref, Medline, Google Scholar
34 : Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry 2008; 63:809–815 Crossref, Medline, Google Scholar
35 : Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: a prospective birth cohort study. Schizophr Bull 2000; 26:287–295 Crossref, Medline, Google Scholar
36 : Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 2004; 161:889–895 Link, Google Scholar
37 : Serologic evidence for prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry 2004; 61:774–780 Crossref, Medline, Google Scholar
38 : Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry 2005; 162:767–773 Link, Google Scholar
39 : Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry 2006; 163:927–929 Link, Google Scholar
40 : No evidence of relation between maternal exposure to herpes simplex virus type 2 and risk of schizophrenia? Am J Psychiatry 2006; 163:2178–2180 Link, Google Scholar
41 : Schizophrenia and influenza. Lancet 1991; 338:116–117 Crossref, Google Scholar
42 : Prenatal exposure to the 1957 influenza epidemic and adult schizophrenia: a follow-up study. Br J Psychiatry 1996; 168:368–371 Crossref, Medline, Google Scholar
43 : Nonaffective psychosis after prenatal exposure to rubella. Am J Psychiatry 2000; 157:438–443 Link, Google Scholar
44 : A.E. Bennett RESEARCH award: prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol Psychiatry 2001; 49:473–486 Crossref, Medline, Google Scholar
45 : Toxoplasma gondii as a risk factor for early-onset schizophrenia: analysis of filter paper blood samples obtained at birth. Biol Psychiatry 2007; 61:688–693 Crossref, Medline, Google Scholar
46 : Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull 2009; 35:631–637 Crossref, Medline, Google Scholar
47 : Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry 2009; 166:1025–1030 Link, Google Scholar
48 : The design of the Prenatal Determinants of Schizophrenia study. Schizophr Bull 2000; 26:257–273 Crossref, Medline, Google Scholar
49 : Toxoplasmosis, in Infections of the Central Nervous System. Edited by Scheld WMWhitley RJDurack DT. Philadelphia, Lippincott-Raven, 1997, pp 785–806 Google Scholar
50
51 : Cytokines: stress and immunity: an overview, in Cytokines: Stress and Immunity. Edited by Plotnikoff NPFaith REMurgo AJGood RA. Boca Raton, Fla, CRC Press, 1999, pp 1–15 Google Scholar
52 : The direct embryotoxicity of immunoglobulin-G fractions from patients with systemic lupus erythematosus. Am J Reprod Immunol 1995; 34:349–355 Crossref, Medline, Google Scholar
53 : Defective corticogenesis and reduction in reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry 1999; 4:145–154 Crossref, Medline, Google Scholar
54 : Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 2003; 23:297–302 Crossref, Medline, Google Scholar
55 : Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res 1997; 24:365–367 Crossref, Medline, Google Scholar
56 : Immune involvement in schizophrenia and autism: etiology, pathology, and animal models. Behav Brain Res 2009; 204:313–321 Crossref, Medline, Google Scholar
57 : Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 1997; 42:1–8 Crossref, Medline, Google Scholar
58 : White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry 2003; 60:443–456 Crossref, Medline, Google Scholar
59 : Cytokine production in chorioamnionitis. J Reprod Immunol 2000; 47:185–196 Crossref, Medline, Google Scholar
60 : Amniotic fluid concentrations of interleukin-1beta, interleukin-6, and TNF-alpha in chorioamnionitis before 32 weeks of gestation: histological associations and neonatal outcome.
61 : Association between –G308A tumor necrosis factor alpha gene polymorphism and schizophrenia. Mol Psychiatry 2001; 6:79–82 Crossref, Medline, Google Scholar
62 : Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann NY Acad Sci 2003; 1003:318–327 Crossref, Medline, Google Scholar
63 : NMDA agonists and antagonists as probes of glutamatergic dysfunction and pharmacotherapies in neuropsychiatric disorders. Harv Rev Psychiatry 1999; 7:125–143 Crossref, Medline, Google Scholar
64 : Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for schizophrenia. Biol Psychiatry 2001; 50:743–749 Crossref, Medline, Google Scholar
65 : Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27:10695–10702 Crossref, Medline, Google Scholar
66 : Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 2005; 10:40–68 Crossref, Medline, Google Scholar
67 : Molecular genetic studies of schizophrenia. Eur J Hum Genet 2006; 14:669–680 Crossref, Medline, Google Scholar
68 : Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron 2000; 25:79–91 Crossref, Medline, Google Scholar
69 : Disrupted-in-schizophrenia 1 and neuregulin 1 are required for the specification of oligodendrocytes and neurones in the zebrafish brain. Hum Mol Genet 2009; 18:391–404 Crossref, Medline, Google Scholar
70 : A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 2005; 7:1167–1178 Crossref, Medline, Google Scholar
71 : Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 2008; 13:173–186 Crossref, Medline, Google Scholar
72 : Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci USA 2005; 102:8627–8632 Crossref, Medline, Google Scholar
73 : Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J Neurosci 2008; 28:6872–6883 Crossref, Medline, Google Scholar
74 : Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci USA 2008; 105:14157–14162 Crossref, Medline, Google Scholar
75 : Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell 2006; 125:127–142 Crossref, Medline, Google Scholar
76 : Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci 2008; 25:4974–4984 Crossref, Google Scholar
77 : Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated DISC1. J Neurosci 2008; 28:10893–10904 Crossref, Medline, Google Scholar
78 : Neural development of the neuregulin receptor ErbB4 in the cerebral cortex and the hippocampus: preferential expression by interneurons tangentially migrating from the ganglionic eminences. Cereb Cortex 2003; 13:252–264 Crossref, Medline, Google Scholar
79 : Axonal neuregulin-1 regulates myelin sheath thickness. Science 2004; 304:700–703 Crossref, Medline, Google Scholar
80 : Enlargement of the lateral ventricles in mutant DISC1 transgenic mice. Mol Psychiatry 2008; 13:115 Crossref, Medline, Google Scholar
81 : A mutation in mouse DISC1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci USA 2008; 105:7076–7081 Crossref, Medline, Google Scholar
82 : Phenotypic characterization of cognition and social behavior in mice with heterozygous versus homozygous deletion of catechol-O-methyltransferase. Neuroscience 2008; 155:1021–1029 Crossref, Medline, Google Scholar
83 : Behavioral phenotypes of DISC1 missense mutations in mice. Neuron 2007; 54:387–402 Crossref, Medline, Google Scholar
84 : Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. Neuroreport 2005; 16:271–275 Crossref, Medline, Google Scholar
85 : Heterozygous neuregulin 1 mice display greater baseline and delta(9)-tetrahydrocannabinol-induced c-Fos expression. Neuroscience 2007; 149:861–870 Crossref, Medline, Google Scholar
86 : Phenotypic characterization of spatial cognition and social behavior in mice with "knockout" of the schizophrenia risk gene neuregulin 1. Neuroscience 2007; 147:18–27 Crossref, Medline, Google Scholar
87 : Disruption to social dyadic interactions but not emotional/anxiety-related behaviour in mice with heterozygous "knockout" of the schizophrenia risk gene neuregulin-1. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:462–466 Crossref, Medline, Google Scholar
88 : Immune signalling in neural development, synaptic plasticity, and disease. Nat Rev Neurosci 2004; 5:521–531 Crossref, Medline, Google Scholar
89 : Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry 2006; 59:929–939 Crossref, Medline, Google Scholar
90 et al: Common variants conferring risk of schizophrenia. Nature 2009; 460:744–747 Crossref, Medline, Google Scholar
91 : Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460:753–757 Crossref, Medline, Google Scholar
92 : Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460:748–752 Crossref, Medline, Google Scholar
93 : Psychiatric Epidemiology: Searching for the Causes of Mental Disorders. New York, Oxford University Press, 2006 Google Scholar
94 : Modern Epidemiology, 3rd ed. Philadelphia, Lippincott Williams & Wilkins, 2008 Google Scholar
95 : Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry 2001; 58:361–367 Crossref, Medline, Google Scholar
96 : Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002; 159:1080–1092 Link, Google Scholar
97 : Decline in the incidence of schizophrenia in Finnish cohorts born from 1954 to 1965. Arch Gen Psychiatry 1999; 56:733–740 Crossref, Medline, Google Scholar
98 : Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386–389 Crossref, Medline, Google Scholar
99 : Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene × environment interaction. Biol Psychiatry 2005; 57:1117–1127 Crossref, Medline, Google Scholar
100 et al: Analysis of 10 independent samples provides evidence for association between schizophrenia and a SNP flanking fibroblast growth factor receptor 2. Mol Psychiatry 2009; 14:30–36 Crossref, Medline, Google Scholar
101 et al: Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet 2008; 40:1053–1055 Crossref, Medline, Google Scholar

