
Elevated Putamen D 2 Receptor Binding Potential in Major Depression With Motor Retardation: An [ 11 C]Raclopride Positron Emission Tomography Study
Abstract
Objective: Several antidepressants raise striatal dopamine, but the role of striatal dopamine during major depressive episodes is unclear. Striatal [ 11 C]raclopride binding potential measured with positron emission tomography is an index of D 2 type receptors and is sensitive to extracellular dopamine levels (higher D 2 binding potential occurs when dopamine is lower). It was hypothesized that putamen D 2 binding potential would be higher during major depressive episodes featuring motor retardation. Method: Drug-free, nonsmoking subjects experiencing a major depressive episode (N=21) underwent [ 11 C]raclopride PET imaging as did 21 healthy age-matched comparison subjects. Motor retardation was measured with the finger tapping test. Results: The depressed subjects exhibiting motor retardation had significantly higher D 2 binding potential in both the left and right putamen than did healthy subjects, and putamen D 2 binding potential correlated significantly with motor speed in the depressed subjects. Conclusions: The results argue that extracellular dopamine is lower in subjects experiencing a major depressive episode that features motor retardation. This depression subtype should preferentially benefit from dopamine-increasing medications and should be targeted in future clinical trials of dopamine reuptake inhibitors.
It is generally believed that pathophysiological processes implicated in major depressive disorder are reversed by antidepressant treatment. Most antidepressants raise monoamines, and the majority of these antidepressants raise serotonin and/or norepinephrine. However, a number of commonly prescribed antidepressants also have dopamine-increasing properties, the most prominent being sertraline, bupropion, and the entire class of monoamine oxidase A and A/B inhibitors (1 – 4) . Sertraline has a moderately high affinity for the dopamine transporter (1 , 2) . Bupropion has its highest known affinity for the dopamine transporter, and the entire class of monoamine oxidase A and A/B inhibitors robustly increase striatal dopamine in animal models (1 , 3 , 4) . Even though these treatments are often used, the role of dopamine during major depressive episodes is not clear.
Pathological putamen dopamine neurotransmission is associated with motor retardation in a number of illnesses such as Parkinson’s disease, multisystem atrophy, and progressive supranuclear palsy (5 , 6) . Motor retardation is frequently a symptom of major depressive episodes (7) , and striatal dopamine may also be low during major depressive episodes when motor retardation is present, since a very indirect measure of brain dopamine—CSF levels of the dopamine metabolite homovanillic acid (HVA)—is often low during major depressive episodes with motor retardation (8 , 9) .
Striatal dopamine concentrations cannot be directly measured in vivo in humans. However, [ 11 C]raclopride positron emission tomography (PET) is a useful noninvasive method for investigating disease processes that may lower extracellular dopamine. [ 11 C]Raclopride is a validated PET radiotracer that is selective for D 2 type receptors with a modest preference for D 2 receptors over D 3 receptors (10 – 12) . The index of D 2 type receptors found with this method, referred to in this article as D 2 receptor binding potential, is inversely proportional to extracellular dopamine levels in animal paradigms of acute and chronic dopamine depletion as well as acute increases in dopamine (13) . Moreover, the D 2 binding potential is elevated in human conditions associated with dopamine loss such as AMPT (alpha-methyl-paratyrosine) treatment (14) and untreated Parkinson’s disease (15 – 17) . The D 2 binding potential is lower after acute increases in dopamine in humans (13) . Thus, [ 11 C]raclopride PET should be useful for investigating abnormalities of striatal dopamine during major depressive episodes.
To our knowledge there have been no studies to date that have used [ 11 C]raclopride PET in depressed subjects. [ 123 I]IBZM single photon emission computed tomography (SPECT) is a similar method because IBZM binds to D 2 type receptors, and the D 2 binding potential found with [ 123 I]IBZM SPECT shows a similar inverse relationship to extracellular dopamine levels (13) . However, all previous investigations of striatal D 2 binding potential during major depressive episodes using [ 123 I]IBZM SPECT are confounded by antidepressant use (18 – 22) . Four (18 – 21) of the five studies recruited subjects who had taken antidepressants within 2 weeks of scanning. This is a critical issue, since large magnitude changes in D 2 binding potential during acute administration of serotonin-increasing medications have been reported in humans (23) . The one study that recruited some drug-free subjects (22) cannot be considered conclusive because it did not compare the drug-free subjects to the healthy subjects. Of the five studies, only one (20) measured both striatal D 2 binding potential and motor speed with neuropsychological testing. This study is also not definitive because most of the subjects were taking antidepressant medication, subjects with bipolar disorder were included, and the effects of age were not addressed. Striatal D 2 binding potential declines with age (24) , and motor speed declines with age (25) . Therefore, controlling for the effect of age on both variables is essential or else a spurious association between the two is possible.
The underlying hypothetical model for the present study was that extracellular dopamine would be low in the putamen during major depressive episodes when motor retardation is present. The evidence supporting this model is that CSF concentration of the dopamine metabolite, HVA, is low during depression with motor retardation (8 , 9) and that low dopamine neurotransmission in the putamen is associated with motor retardation in several other diseases (5 , 6) . Extracellular striatal dopamine cannot be measured directly in vivo in humans; however, the D 2 binding potential found with [ 11 C]raclopride PET is inversely correlated with extracellular dopamine levels (13 , 14 , 16) . Therefore, our specific hypothesis was that putamen D 2 binding potential would be elevated in depressed subjects exhibiting motor retardation.
Method
Subjects
Twenty-one depressed subjects (nine men and 12 women; mean age=35 years [SD=10]) experiencing a current major depressive episode and 21 age-matched healthy subjects (nine men and 12 women; mean age=34 years [SD=11]) were recruited. All subjects were physically healthy, nonsmoking, and had no history of neurotoxin or neuroleptic use. Subjects who smoked were excluded because cigarette smoking in humans acutely lowers [ 11 C]raclopride PET measurement of D 2 binding potential in ventral striatum (26) . All subjects received a urine drug screen on the day of the [ 11 C]raclopride PET scan.
Diagnoses of major depressive episode secondary to major depressive disorder were determined with the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID) and confirmed by a psychiatrist (J.H.M.). For subjects experiencing a major depressive episode, the minimum severity for study enrollment was a score of 17 on the 17-item Hamilton Depression Rating Scale. Additional clinical details are presented in Table 1 . The SCID also was used to rule out comorbid axis I disorders in the depressed subjects. Subjects whose major depressive episode included psychotic symptoms were also excluded. All depressed subjects received blood tests (thyroid function, electrolytes, complete blood cell count) to rule out medical causes of disturbed mood.

Healthy subjects were screened with the SCID to rule out any axis I disorders. All subjects were screened to rule out axis II disorders using the SCID for axis II disorders (27) .
For each subject, written consent was obtained after the procedures had been fully explained. The study and recruitment procedures were approved by the research ethics board for human subjects at the Centre for Addiction and Mental Health.
PET Scanning
For each PET scan, 370 MBq of [ 11 C]raclopride was administered intravenously as a bolus. Frames were acquired as follows: five frames of 1 minute, 20 frames of 2 minutes, and three frames of 5 minutes. [ 11 C]Raclopride was of high radiochemical purity (>95%) and high specific activity (32 GBq/μmol [SD=15] at the time of injection). PET images were obtained using a GEMS 2048–15B camera (intrinsic resolution=5.5 mm full width at half maximum). All images were corrected for attenuation using a 68 Ge transmission scan and reconstructed by filtered back projection (Hanning filter).
To derive D 2 binding potential, the simplified reference tissue model (12) was applied to time activity curve data from [ 11 C]raclopride PET scanning (28) . The binding potential found with this noninvasive method represents the ratio of specifically bound radiotracer to radiotracer in free and nonspecific compartments at equilibrium state. This binding potential is equal to (1/V 2 )*(B max /K d ) (12) . B max represents receptor density, K d is the dissociation constant (which is inversely proportional to affinity), and V 2 is the ratio of free and nonspecifically bound radiotracer in tissue relative to plasma concentration if they were at an equilibrium state (which is an index of free and nonspecific binding in tissue).
For the region of interest method, each subject underwent MRI scanning (GE Signa 1.5-T scanner, spin-echo sequence, T 1 weighted image; x, y, z voxel dimensions 0.78, 0.78, and 3 mm, respectively). Regions of interest were drawn on MRI scans that were coregistered to each summated [ 11 C]raclopride PET image using a mutual information algorithm (29) . The location of the region of interest was verified by visual assessment of the region of interest upon the summated [ 11 C]raclopride PET image. Left and right caudate and putamen regions of interest were drawn on two adjacent slices and the cerebellum region of interest (the reference region) was drawn bilaterally on two adjacent slices as described previously (30) .
D 2 binding potential was found in individual voxels using the basis function application of the simplified reference tissue model (11) . Images were spatially normalized to a common brain shape using statistical parametric mapping (31) and ligand specific templates (32) .
Neuropsychological Testing
All healthy subjects and 19 of 21 depressed subjects completed neuropsychological testing that covered several domains of function ( Table 2 ). The finger tapping test was selected as a measure of motor speed for several reasons. First, this test is sensitive to motor retardation in a number of illnesses, including major depressive disorder (7 , 25) . Second, the test is extremely reliable because the test itself involves at least five consecutive measures of finger tapping (25) . Third, test performance has been associated with disturbances in dopaminergic neurotransmission in several conditions and illnesses, including neuronal loss of aging (24) , [ 18 F]-dopa uptake in Parkinson’s disease (33) , and dopamine transporter binding potential during major depressive episodes (34) . All neuropsychological tests were administered according to standardized procedures and blind to measurement of D 2 binding potential.

Statistical Methods
First, we determined the relationship between D 2 binding potential and age in each striatal region using an analysis of covariance (ANCOVA) so that we could control for age effects in our region of interest analyses. It is well known that striatal D 2 receptor density declines with age (24) . For other region of interest analyses, the effect of age was removed from the D 2 binding potential using the slope of the linear regression between regional D 2 binding potential and age such that the regional D 2 binding potential for each subject was normalized to the D 2 binding potential for a 30-year-old subject. The spatial extent of the relationship between age and D 2 binding potential in striatum was assessed using an analysis of covariance for each voxel.
Given that there have been no investigations of striatal D 2 binding potential in drug-free, nonsmoking, depressed subjects, the next step was comparing striatal D 2 binding potential between depressed and healthy subjects using an analysis of variance for each region of interest. The spatial extent of this relationship in the striatum was also assessed using an analysis of variance for each voxel to assess the effect of diagnosis while controlling for age.
To test the primary hypothesis, a subgroup of depressed subjects exhibiting motor retardation was generated. First, the variance related to demographic variables of age and gender was removed from the measurement of motor retardation using the slopes of linear regression to remove the effects of these variables, normalizing the scores to a 30-year-old male subject. We have used these approaches previously (34 , 35) . An important reason to use this method is that the potential for a spurious correlation between D 2 binding potential and motor retardation based upon the common covariation of each variable with age is removed. Next, the subgroup of depressed subjects exhibiting motor retardation was found by selecting those subjects with motor speed less than or equal to the median finger tapping test score of all depressed subjects.
We then tested the primary hypothesis by comparing putamen D 2 binding potential between the subgroup of depressed subjects with motor retardation and healthy subjects using an analysis of variance (ANOVA). The spatial extent of this relationship in the striatum was also assessed using an analysis of variance for each voxel to assess the effect of group (while controlling for age). We also examined the relationship between motor speed and putamen D 2 binding potential in the depressed group using Pearson correlation coefficients, since this information is also relevant to the primary hypothesis. The spatial extent of this relationship in the striatum was also assessed using an analysis of covariance for each voxel to assess the effect of motor speed (while controlling for age).
Last, we investigated the relationship between striatal D 2 binding potential and other neuropsychological and clinical measures using Pearson correlation coefficients. In these analyses, for neuropsychological measurements in which there are known relationships to age or gender, the variance related to these demographic variables was removed from the measurement using the slopes of linear regression for the effects of these variables, normalizing scores to a 30-year-old male subject. With the exception of the finger tapping test, the correlations found were not directly related to the primary hypothesis and were not corrected for number of comparisons, so they should be viewed as exploratory.
Results
Effect of Age and Gender Upon Striatal D 2 Binding Potential
We found that in each region of interest (whole striatum, left and right caudate, left and right putamen), D 2 binding potential declined with age (ANCOVA F=11.0–21.0, df=1, 40, p≤0.002). The voxel analysis showed that D 2 binding potential within the entire striatum correlated significantly with age (left striatum: N=1765 suprathreshold voxels, p<0.001 [corrected for cluster size]; right striatum: N=1332 suprathreshold voxels, p<0.001 [corrected for cluster size]). There was no effect of gender upon striatal D 2 binding potential (ANCOVA F=2.3, df=1, 39, p=0.13).
Effect of Major Depressive Episode on Striatal D 2 Binding Potential
As seen in Figure 1 , D 2 binding potential in depressed subjects was significantly elevated in every striatal region (ANOVA F=4.99–10.23, df=1, 40, p=0.03–0.002; magnitude: 6% to 8%). Somewhat more motor retardation was seen in the depressed subjects (mean=42.6 taps in 10 seconds) than in the healthy subjects (mean=47.0 taps in 10 seconds) (t=1.8, df=38, p=0.08). In the voxel analysis, D 2 binding potential in the depressed subjects was elevated in the entire striatum ( Figure 2 ).
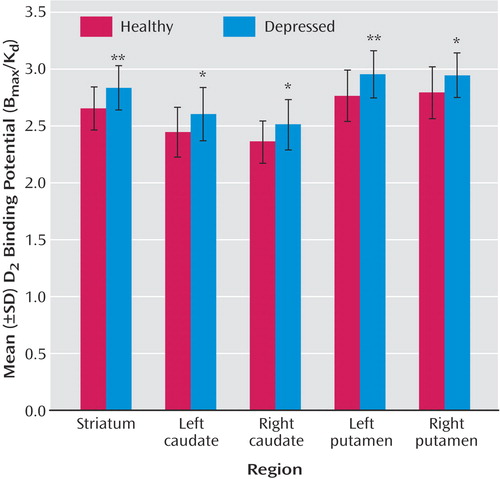
a D 2 binding potential values were normalized to a 30-year-old subject using the slope of the age-related decline. Between-group differences assessed by means of independent sample t tests.
* p≤0.05. ** p≤0.005.

a After correction for cluster significance, ANCOVA with age as a covariate revealed two significant clusters depicting the effect of a major depressive episode (left striatum: N=1278 suprathreshold voxels, p<0.001 [corrected for cluster size]; right striatum: N=1020 suprathreshold voxels, p<0.001 [corrected for cluster size]). Threshold for display, p≤0.05.
Striatal D 2 Binding Potential in Depressed Subjects With Motor Retardation
The subgroup of depressed subjects with motor retardation (N=10) exhibited significantly greater motor retardation (mean=37.3 taps in 10 seconds) than did the healthy subjects (mean=47.0 taps in 10 seconds) (t=3.37, df=29, p=0.002). No differences between depression subgroups was found with respect to age, gender, or number of depressive episodes.
As seen in Figure 3 , the depressed subgroup with motor retardation had significantly higher D 2 binding potential in each putamen region than the healthy subjects (left putamen: F=10.54, df=1, 29, p=0.003 [magnitude 10%]; right putamen: F=6.86, df=1, 29, p=0.01 [magnitude 8%]). In the voxel analysis, D 2 binding potential was elevated throughout the striatum in the depressed subjects with motor retardation ( Figure 4 ).

a D 2 binding potential values were normalized to a 30-year-old subject using the slope of the age-related decline. Differences between the healthy group and the depressed subjects with motor retardation assessed by means of independent sample t tests.
* p≤0.05. ** p≤0.01. *** p≤0.005.

a After correction for cluster significance, ANCOVA with age as a covariate revealed two significant clusters depicting the effect of a major depressive episode with motor retardation (left striatum: N=1277 suprathreshold voxels, p<0.001 [corrected for cluster size]; right striatum: N=1174 suprathreshold voxels, p<0.001 [corrected for cluster size]). Threshold for display, p≤0.05.
Analysis of the entire group of depressed subjects showed a significant correlation between the measurement of motor speed with the finger tapping test and the D 2 binding potential in the left putamen (r=–0.50, df=17, p=0.03) and right putamen (r=–0.50, df=17, p=0.03) ( Figure 5 ). In addition, motor speed measured with the finger tapping test tended to be associated with D 2 binding potential in the left caudate (r=–0.44, df=17, p=0.06) and the right caudate (r=–0.43, df=17, p=0.07). It was evident that the D 2 binding potential among regions was significantly correlated within the depressed individuals: the various correlations in age-normalized D 2 binding potential between the left and right caudate and left and right putamen ranged from 0.52 to 0.79 (p=0.02 to p≤0.001). Similar results were found in the voxel analysis: the D 2 binding potential in the entire striatum was correlated with finger tapping scores ( Figure 6 ).

a In order to reduce variance related to age and gender, individual putamen D 2 binding potential values were normalized to a 30-year-old subject using the slope of the age-related decline. Individual finger tapping scores were normalized similarly to a 30-year-old male subject. The correlation was significant (r=–0.53, df=17, p=0.02).

a After correction for cluster significance, ANCOVA with age as a covariate revealed two significant clusters depicting the effect of motor speed (left striatum: N=1665 suprathreshold voxels, p<0.001 [corrected for cluster size]; right striatum: N=1523 suprathreshold voxels, p<0.001 [corrected for cluster size]). Threshold for display, p≤0.05.
Secondary Neuropsychological and Clinical Measures and Regional D 2 Binding Potential in Depressed Subjects
Since the D 2 binding potential among regions was correlated within individuals, the correlations between other neuropsychological measures and whole striatal D 2 binding potential was examined first ( Table 2 ). There were no significant correlations between individual neuropsychological tests and regional D 2 binding potential in either group (except for the finger tapping test). In addition, post hoc analyses of clinical measures showed that no other clinical variable (including number of previous episodes, length of current episode, history of antidepressant treatment) was significantly correlated with regional D 2 binding potential.
Discussion
This is the first study to investigate striatal D 2 binding potential in medication-free, nonsmoking depressed subjects. The main findings were that caudate and putamen D 2 binding potential were elevated in the depressed group as compared with the healthy group, and that putamen D 2 binding potential was most significantly elevated in the depressed subgroup with motor retardation. Moreover, there was a significant correlation between putamen D 2 binding potential and motor speed in the depressed group. These findings are important for understanding the pathophysiology of depression because they represent a striatal dopaminergic disturbance during depression that is most prominent when motor retardation is present. This has implications for the monoamine theory of depression. In addition, these findings have important implications for treatment, since this dopamine abnormality may be targeted by dopamine-increasing antidepressants.
The best interpretation of these findings is that putamen dopamine is low in depression with motor retardation. Elevations in D 2 binding potential measured with [ 11 C]raclopride PET may occur when dopamine is low (13 , 14) , and the most significant elevations in putamen D 2 binding potential were observed in the depressed subjects with motor retardation ( Figure 3 ). These results are consistent with previous reports of lower CSF metabolite of dopamine in depressed subjects with motor retardation (8 , 9) and the general disease model associating motor retardation with impaired dopaminergic neurotransmission in the putamen (5 , 6) . These results are also consistent with our previous finding of lower dopamine transporter binding potential in a similar group of medication-free depressed subjects (34) , since dopamine transporter density can decrease during chronic dopamine-depleting paradigms (36 , 37) . The other investigation of striatal dopamine transporter in medication-free depressed subjects sampled those with seasonal affective disorder and also found lower striatal dopamine transporter binding potential (38) .
The results of the present study have important implications for understanding the neurochemistry of major depressive disorder. In many versions of the monoamine theory of depression, major depressive disorder is viewed as having a pathology of homogenous monoamine loss. A contrasting view is that specific monoamines are lowest when specific symptoms of depression are most severe (35) . Although putamen D 2 binding potential was elevated in the depressed group as compared with the healthy group, the data in Figure 3 show that the elevation in putamen (and striatal) D 2 binding potential was driven by the depressed group with motor retardation. Since putamen D 2 binding potential is highest in the depressed subjects with motor retardation and since measurement of D 2 binding potential with [ 11 C]raclopride PET is inversely correlated with extracellular dopamine levels (13 , 14 , 16) , the results argue for heterogeneous monoamine loss in depression (with greater specific monoamine loss when specific symptoms are more severe).
The interpretation that extracellular dopamine is low in depressed subjects with motor retardation has substantial implications for treatment because it argues for a preferential response in this subtype to antidepressants that induce sustained dopamine agonist effects such as dopamine reuptake inhibitors (i.e., bupropion and sertraline) and monoamine oxidase A and A/B inhibitors (1 – 4) . Moreover, it is likely that further dopamine reuptake inhibitor development will occur, since current treatments are unlikely to be optimally therapeutic at this target site. For example, in contrast to the 80% or greater occupancy of serotonin transporters typically found at therapeutic dosing of serotonin reuptake inhibitors (35) , dopamine transporter blockade with sustained-release bupropion has been estimated between 0% and 26% (39 – 41) . The other commonly used antidepressant with dopamine reuptake inhibitor properties, sertraline, has approximately two orders of magnitude higher affinity for the serotonin transporter over the dopamine transporter (1 , 2) , and it is probable that a closer affinity ratio between these two sites would be more optimal for therapeutic effect. A related class, dopamine-releasing medications, is available, but their mechanism is dependent upon intact dopamine storage (42) , making them less clinically relevant. It is well known that serotonin-releasing medications like d -fenfluramine are not therapeutically relevant for treating depression. It is also theoretically possible that dopamine agonists that stimulate dopamine receptors in a similar manner to dopamine itself could also prove to be therapeutically useful in treating motor retardation during major depression. The results of the present study argue for 1) the use of existing dopamine-increasing medications to treat depression with motor retardation and 2) the development of antidepressants with higher dopamine reuptake inhibitor blockade for depression with motor retardation.
Striatal D 2 binding potential was 9% higher in the putamen of depressed subjects with motor retardation as compared with the healthy subjects. Although this elevation of putamen D 2 binding potential in depressed subjects with motor retardation was significantly higher than healthy subjects, it is less than what occurs in early untreated Parkinson’s disease (estimates with [ 11 C]raclopride PET measurement typically range from 7% to 30% [ 15 – 17 ], averaging at 20%) or after a severe acute dopamine depletion with alpha-methyl-paratyrosine (18% [14] ). This is not surprising: the loss of dopamine neurons in Parkinson’s disease is generally considered a large magnitude effect, and the version of the alpha-methyl-paratyrosine challenge used by Verhoeff et al. (14) was a drastic paradigm tolerated for only a brief period under inpatient supervision.
This study has limitations inherent in the measurement of D 2 binding potential that are common with in vivo imaging studies of disease states. Striatal D 2 binding potential as measured with [ 11 C]raclopride reflects a combination of D 2 receptor density, affinity, and accessibility of the radioligand to D 2 receptor sites. While D 2 binding potential can certainly increase under conditions of acute and chronic dopamine depletion (13 , 14) , there are a number of other potential explanations for a greater striatal D 2 binding potential. Therefore, our interpretation of low extracellular dopamine is also dependent upon other dopamine-related investigations in major depressive disorder. We did not measure hedonia, since neuropsychological measurements of hedonia need to be developed for major depressive disorder. Self-report of hedonia is potentially prone to biases of severely negativistic thinking (where every experience is viewed pessimistically, resulting in underreporting) and anxiety (where people are hesitant to incorrectly report and therefore underreport). Some PET study designs also measure D 2 binding potential after dopamine depletion and assume that the dopamine depletion would result in similar final dopamine levels in patients and healthy comparison subjects (13) . This assumption seems contradictory to our hypothesized model of abnormally regulated extracellular dopamine levels in depression, so we did not apply a dopamine-depleting paradigm. Our previous work found that the striatal dopamine transporter binding potential is lower in depression (34) —as would be expected during a dopamine-lowering process (36 , 37) —and when it is lowest, it is actually protective against motor retardation. We surmised that a lower striatal dopamine transporter binding potential would result in less clearance of extracellular dopamine and would be protective against a different dopamine-lowering process. Promising mechanisms for a major dopamine lowering process include dysregulations of enzymes of dopamine synthesis or dopamine metabolism, and these will be studied in future research.
This study was the first investigation of striatal D 2 binding potential in drug-free, nonsmoking, currently depressed subjects with a reasonably large patient group size. We found that depressed subjects with motor retardation had a significantly elevated striatal D 2 binding potential relative to healthy volunteers. Moreover, among depressed subjects, higher striatal D 2 binding potential was correlated with more severe motor retardation. The best explanation for these findings is that lower extracellular dopamine occurs in depressed subjects with motor retardation, since this explanation is consistent with reports of low CSF HVA in depression with motor retardation (8 , 9) , low dopamine transporter binding potential in depression (34) , and an association between putamen dopaminergic pathology and motor retardation in other disease models (5 , 6 , 15) . Low dopamine pathology in depression with motor retardation has important implications for the treatment of motor retardation in depression. This subtype of depression should benefit from dopamine-increasing medications, and future clinical trials of dopamine reuptake inhibitor treatments should be conducted in depression with motor retardation.
1. Tatsumi M, Groshan K, Blakely RD, Richelson E: Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 1997; 340:249–258Google Scholar
2. Owens MJ, Knight DL, Nemeroff CB: Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry 2001; 50:345–350Google Scholar
3. Colzi A, d’Agostini F, Kettler R, Borroni E, Da Prada M: Effect of selective and reversible MAO inhibitors on dopamine outflow in rat striatum: a microdialysis study. J Neural Transm Suppl 1990; 32:79–84Google Scholar
4. Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW: Effects of selective monoamine oxidase inhibitors on the in vivo release and metabolism of dopamine in the rat striatum. J Neurochem 1990; 55:981–988Google Scholar
5. Kim YJ, Ichise M, Ballinger JR, Vines D, Erami SS, Tatschida T, Lang AE: Combination of dopamine transporter and D2 receptor SPECT in the diagnostic evaluation of PD, MSA, and PSP. Mov Disord 2002; 17:303–312Google Scholar
6. Kish SJ, Shannak K, Hornykiewicz O: Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease: pathophysiologic and clinical implications. N Engl J Med 1988; 318:876–880Google Scholar
7. Raskin A, Friedman AS, DiMascio A: Cognitive and performance deficits in depression. Psychopharmacol Bull 1982; 18:196–202Google Scholar
8. Korf J, van Praag HM: Retarded depression and the dopamine metabolism. Psychopharmacologia 1971; 19:199–203Google Scholar
9. Post RM, Kotin J, Goodwin FK, Gordon EK: Psychomotor activity and cerebrospinal fluid amine metabolites in affective illness. Am J Psychiatry 1973; 130:67–72Google Scholar
10. Seeman P, Van Tol HH: Dopamine receptor pharmacology. Curr Opin Neurol Neurosurg 1993; 6:602–608Google Scholar
11. Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ: Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. Neuroimage 1997; 6:279–287Google Scholar
12. Lammertsma AA, Hume SP: Simplified Reference Tissue Model for PET Receptor Studies. 1996; 4:153–158Google Scholar
13. Laruelle M: Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 2000; 20:423–451Google Scholar
14. Verhoeff NP, Kapur S, Hussey D, Lee M, Christensen B, Psych C, Papatheodorou G, Zipursky RB: A simple method to measure baseline occupancy of neostriatal dopamine D2 receptors by dopamine in vivo in healthy subjects. Neuropsychopharmacology 2001; 25:213–223Google Scholar
15. Brooks DJ, Ibanez V, Sawle GV, Playford ED, Quinn N, Mathias CJ, Lees AJ, Marsden CD, Bannister R, Frackowiak RS: Striatal D2 receptor status in patients with Parkinson’s disease, striatonigral degeneration, and progressive supranuclear palsy, measured with 11C-raclopride and positron emission tomography. Ann Neurol 1992; 31:184–192Google Scholar
16. Rinne UK, Laihinen A, Rinne JO, Nagren K, Bergman J, Ruotsalainen U: Positron emission tomography demonstrates dopamine D2 receptor supersensitivity in the striatum of patients with early Parkinson’s disease. Mov Disord 1990; 5:55–59Google Scholar
17. Sawle GV, Playford ED, Brooks DJ, Quinn N, Frackowiak RS: Asymmetrical pre-synaptic and post-synpatic changes in the striatal dopamine projection in dopa naive parkinsonism: diagnostic implications of the D2 receptor status. Brain 1993; 116(pt 4):853–867Google Scholar
18. D’Haenen HA, Bossuyt A: Dopamine D2 receptors in depression measured with single photon emission computed tomography. Biol Psychiatry 1994; 35:128–132Google Scholar
19. Klimke A, Larisch R, Janz A, Vosberg H, Muller-Gartner HW, Gaebel W: Dopamine D2 receptor binding before and after treatment of major depression measured by [123I]IBZM SPECT. Psychiatry Res 1999; 90:91–101Google Scholar
20. Shah PJ, Ogilvie AD, Goodwin GM, Ebmeier KP: Clinical and psychometric correlates of dopamine D2 binding in depression. Psychol Med 1997; 27:1247–1256Google Scholar
21. Parsey RV, Oquendo MA, Zea-Ponce Y, Rodenhiser J, Kegeles LS, Pratap M, Cooper TB, Van Heertum R, Mann JJ, Laruelle M: Dopamine D(2) receptor availability and amphetamine-induced dopamine release in unipolar depression. Biol Psychiatry 2001; 50:313–322Google Scholar
22. Ebert D, Feistel H, Loew T, Pirner A: Dopamine and depression: striatal dopamine D2 receptor SPECT before and after antidepressant therapy. Psychopharmacology (Berl) 1996; 126:91–94Google Scholar
23. Smith GS, Dewey SL, Brodie JD, Logan J, Vitkun SA, Simkowitz P, Schloesser R, Alexoff DA, Hurley A, Cooper T, Volkow ND: Serotonergic modulation of dopamine measured with [11C]raclopride and PET in normal human subjects. Am J Psychiatry 1997; 154:490–496Google Scholar
24. Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, Hitzemann R, Smith G, Logan J: Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry 1998; 155:344–349Google Scholar
25. Spreen O, Strauss E: A Compendium of Neuropsychological Tests. New York, Oxford University Press, 1998Google Scholar
26. Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA: Smoking-induced ventral striatum dopamine release. Am J Psychiatry 2004; 161:1211–1218Google Scholar
27. Blais MA, Norman DK: A psychometric evaluation of the DSM-IV personality disorder criteria. J Personal Disord 1997; 11:168–176Google Scholar
28. Burger C, Buck A: Requirements and implementation of a flexible kinetic modelling tool. J Nucl Med 1997; 38:1818–1823Google Scholar
29. Studholme C, Hill D, Hawkes D: An overlap invariant entropy measure of 3D medical image alignment. Pattern Recognition 1999; 32:71–86Google Scholar
30. Kapur S, Zipursky R, Jones C, Remington G, Houle S: Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000; 157:514–520Google Scholar
31. Ashburner J, Friston KJ: Nonlinear spatial normalization using basis functions. Hum Brain Mapp 1999; 7:254–266Google Scholar
32. Meyer JH, Gunn RN, Myers R, Grasby PM: Assessment of spatial normalization of PET ligand images using ligand-specific templates. Neuroimage 1999; 9:545–553Google Scholar
33. Pal P, Lee C, Samii A, Schulzer M, Stoessl A, Mak E, Wudel J, Dobko T, Tsui J: Alternating two finger tapping with contralateral activation is an objective measure of clinical severity in Parkinson’s disease and correlates with PET [18F]-DOPA Ki. Parkinsonism Relat Dis 2001; 7:305–309Google Scholar
34. Meyer JH, Kruger S, Wilson AA, Christensen BK, Goulding VS, Schaffer A, Minifie C, Houle S, Hussey D, Kennedy SH: Lower dopamine transporter binding potential in striatum during depression. Neuroreport 2001; 12:4121–4125Google Scholar
35. Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N, Goulding VS, Kennedy J, Wilson AA: Brain serotonin transporter binding potential measured with [11C]DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry 2004; 61:1271–1279Google Scholar
36. Kilbourn MR, Sherman PS, Pisani T: Repeated reserpine administration reduces in vivo [18F]GBR 13119 binding to the dopamine uptake site. Eur J Pharmacol 1992; 216:109–112Google Scholar
37. Gordon I, Weizman R, Rehavi M: Modulatory effect of agents active in the presynaptic dopaminergic system on the striatal dopamine transporter. Eur J Pharmacol 1996; 298:27–30Google Scholar
38. Neumeister A, Willeit M, Praschak-Rieder N, Asenbaum S, Stastny J, Hilger E, Pirker W, Konstantinidis A, Kasper S: Dopamine transporter availability in symptomatic depressed patients with seasonal affective disorder and healthy controls. Psychol Med 2001; 31:1467–1473Google Scholar
39. Meyer JH, Goulding VS, Wilson AA, Hussey D, Christensen BK, Houle S: Bupropion occupancy of the dopamine transporter is low during clinical treatment. Psychopharmacology (Berl) 2002; 163:102–105Google Scholar
40. Kugaya A, Seneca NM, Snyder PJ, Williams SA, Malison RT, Baldwin RM, Seibyl JP, Innis RB: Changes in human in vivo serotonin and dopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology 2003; 28:413–420Google Scholar
41. Learned-Coughlin SM, Bergstrom M, Savitcheva I, Ascher J, Schmith VD, Langstrom B: In vivo activity of bupropion at the human dopamine transporter as measured by positron emission tomography. Biol Psychiatry 2003; 54:800–805Google Scholar
42. Butcher SP, Liptrot J, Aburthnott GW: Characterisation of methylphenidate and nomifensine induced dopamine release in rat striatum using in vivo brain microdialysis. Neurosci Lett 1991; 122:245–248Google Scholar

