
Genome-Wide Association Study of Major Recurrent Depression in the U.K. Population
Abstract
Objective
Studies of major depression in twins and families have shown moderate to high heritability, but extensive molecular studies have failed to identify susceptibility genes convincingly. To detect genetic variants contributing to major depression, the authors performed a genome-wide association study using 1,636 cases of depression ascertained in the U.K. and 1,594 comparison subjects screened negative for psychiatric disorders.
Method
Cases were collected from 1) a case-control study of recurrent depression (the Depression Case Control [DeCC] study; N=1346), 2) an affected sibling pair linkage study of recurrent depression (probands from the Depression Network [DeNT] study; N=332), and 3) a pharmacogenetic study (the Genome-Based Therapeutic Drugs for Depression [GENDEP] study; N=88). Depression cases and comparison subjects were genotyped at Centre National de Génotypage on the Illumina Human610-Quad BeadChip. After applying stringent quality control criteria for missing genotypes, departure from Hardy-Weinberg equilibrium, and low minor allele frequency, the authors tested for association to depression using logistic regression, correcting for population ancestry.
Results
Single nucleotide polymorphisms (SNPs) in BICC1 achieved suggestive evidence for association, which strengthened after imputation of ungenotyped markers, and in analysis of female depression cases. A meta-analysis of U.K. data with previously published results from studies in Munich and Lausanne showed some evidence for association near neuroligin 1 (NLGN1) on chromosome 3, but did not support findings at BICC1.
Conclusions
This study identifies several signals for association worthy of further investigation but, as in previous genome-wide studies, suggests that individual gene contributions to depression are likely to have only minor effects, and very large pooled analyses will be required to identify them.
Depression is a common and disabling condition that is a leading cause of disability, responsible for a substantial proportion of absence from work, impaired quality of life, poor physical health, and deaths by suicide (1–3). Increasing our understanding of the etiology of depression is therefore an important priority for medical research.
Family and twin studies indicate a substantial role of genetic factors in the etiology of depression. However, attempts to quantify the genetic contribution to depression have led to estimates of heritability ranging from 17% to 75%, with an average of 37% (2, 4, 5).These discrepant estimates point to a heterogeneity in depression and can be explained by several features associated with heritability. First, cases of depression ascertained through clinical contact tend to have stronger genetic contribution than cases diagnosed by lay interviewers in the general population (4, 5). Second, recurrent depression is more heritable than a single episode (4, 6). Third, more reliable assessment of depression is associated with high heritability, suggesting that heritability is often underestimated due to measurement error (7–9). These lines of evidence all indicate a strong genetic contribution to severe and recurrent depression when it is reliably diagnosed in the context of clinical practice.
While the existence of genetic vulnerability to depression is well-established, progress in the identification of its molecular basis has been slow. Functional candidate gene studies have identified few replicable associations (10), and genome-wide linkage studies have yielded suggestive rather than conclusive results (11). Two genome-wide association studies (GWAS) have been published to date. A population-ascertained study of unipolar depression from the Netherlands, using healthy subjects selected for low liability to depression, detected suggestive evidence for a role of genetic variants in the PCLO gene, but replication studies were inconsistent (12). A second genome-wide study analyzed two cohorts of recurrent depression cases from Munich and Lausanne, along with healthy comparison subjects screened to exclude depression and anxiety, but found no evidence for genetic variants associated with depression (13). These studies indicate that the genetic liability to depression is likely to involve multiple genetic variants of weak effects.
Further studies are needed to identify genetic variants contributing to depression. We performed a genome-wide association study for depression in an extensive U.K. case-control collection. To optimize the power to detect associated variants, we focused on depression cases with the strongest genetic contribution: those with reliably assessed, clinically ascertained recurrent unipolar depression. To improve further power, these depression cases were compared with subjects screened for absence of psychiatric disorders. We also performed a meta-analysis of results from this study and from the Munich and Lausanne depression GWAS (13) to assess evidence for association to depression in over 3,000 cases.
Method
Samples
In the U.K. study, depression cases and comparison subjects were drawn from two studies of recurrent depression and a pharmacogenetic study of antidepressant response using similar methods of case definition and collected by the same clinical team. The studies were approved by the local ethical committees and informed written consent was obtained from all participants.
The Depression Case Control (DeCC) study consists of 1,346 cases (69.3% women) of recurrent depression fulfilling DSM-IV or ICD-10 criteria of at least moderate severity, ascertained from three U.K. centers (London, Cardiff, Birmingham) (14). The mean age at onset was 22.9 years (SD=10.8 years). Probands from the Depression Network (DeNT) affected sibling pair linkage study (15, 16) are cases of recurrent depression of at least moderate severity. The 332 U.K.-ascertained cases included in this analysis were primarily women (75.3%), with mean age at illness onset of 23.0 years (SD=10.2 years). The pharmacogenetic study, the Genome-Based Therapeutic Drugs for Depression (GENDEP) study, comprises subjects who have been investigated while in an episode of depression of at least moderate severity (17, 18). The majority of the 88 U.K. cases included in this analysis were female (63.6%), with mean age at onset of 26.4 years (SD=11.3). In contrast to the DeCC and DeNT studies, GENDEP subjects were not required to have had two or more episodes of depression, although 68 cases (77.3%) were recurrent.
All cases were interviewed with the Schedules for Clinical Assessment in Neuropsychiatry (SCAN) (19), focusing on their worst and second-worst episodes of depression in the DeCC and DeNT studies and on their current episode in GENDEP (see Farmer et al. [15] for full details), with study coordinators for all three studies (and the Munich study described below) trained by one investigator (A.E.F.), giving a homogenous cohort of depression cases. Subjects were excluded if there was a history or family history of schizophrenia or bipolar disorder or if mood symptoms were related to alcohol or substance misuse.
The comparison subjects were 1,288 individuals contacted via the Medical Research Council general practice research framework and screened using a composite index of depressive and anxiety symptoms (20); they were interviewed by telephone using the Past History Schedule (21). Of these subjects, 58.4% were women, with mean age of 47.24 years (range=20–69). A further 457 healthy volunteers (61.4% female) were staff or students of King's College London, again screened for mental health using the Past History Schedule.
Meta-analysis was performed with the U.K. study, a clinical cohort of recurrent depression cases from Munich, and a population-based study in Lausanne with a psychiatric component, as analyzed previously (13). Cases from Munich (N=926) were assessed in a similar manner to the DeCC and DeNT study subjects, with healthy comparison subjects screened for absence of anxiety and mood disorders (N=866) (22). The Lausanne genome-wide association study comprised 492 subjects meeting DSM-IV criteria for recurrent major depression and 1,052 unaffected subjects; these were selected from the Caucasians of a community survey carried out in the city of Lausanne, Switzerland. The methods and the assessment of subjects (based on the Diagnostic Interview for Genetic Studies) have been extensively described (23).
Genotyping
Genotyping for the U.K. depression cases and comparison subjects was performed using the Illumina HumanHap610-Quad BeadChips by the Centre National de Génotypage (CNG), Evry, France. Barcoded DNA samples were received by the CNG DNA banking facility in standard tubes together with the sample information. All DNA samples were subjected to stringent quality control, and processing was carried out under full Laboratory Information Management System (LIMS) control. Confirmatory genotyping of single nucleotide polymorphisms (SNPs) showing suggestive evidence of association was performed to confirm allele-calling and to provide verification of genotypes for imputed SNPs; these genotypes were analyzed separately in testing for association but had higher levels of missing genotypes, limiting their power to detect association.
Quality Control
Stringent quality control procedures were applied to individual and SNP data. Individuals were excluded if their genotypic data showed a missing rate >1%, abnormal heterozygosity, a sex assignment that conflicted with phenotypic data, if they were related (up to second degree) with other study members, or were of non-European ancestry. SNPs with minor allele frequency (<1%) or showing departure from Hardy-Weinberg equilibrium (p<1×10–5) were excluded. Principal component analysis was performed using EIGENSTRAT (24) after quality control procedures, and the two principal components showing significant differences in ancestry between depression cases and comparison subjects were used as covariates in association testing. Further details of quality control are given in the data supplement that accompanies the online version of this article.
Statistical Analysis
The primary test for association between SNPs and depression was logistic regression, including ancestry principal components, assuming a log-additive genetic model. Additional analyses were performed using the Cochran Armitage trend test and logistic regression assuming a full genotype model (2 df) and dominant and recessive models. Analyses were carried out for all depression cases and separately for male and female cases. The genomic control parameter γ was calculated for each analysis to assess inflation due to population stratification (25). Analysis was implemented using PLINK v1.05 (26) and R (www.r-project.org).
Two stringent thresholds of significance were used: genome-wide significance (p<5×10–8 [27, 28]) and suggestive significance (p<5×10–6). This study has a power of 86% at suggestive level of significance (and power of 58% at genome-wide significance) to detect an association explaining 0.75% of variance, assuming that cases account for the upper 10% of the depression liability and screened comparison subjects the lower 70%. For genetic regions where any SNP achieved suggestive evidence for association, follow-up analyses were performed. Genotypes at HapMap SNPs in 200-kb regions flanking the most highly associated SNP were imputed and analyzed using MACH version 1.0 (29) to determine whether stronger associations could be identified in the region. Haplotype analysis of genotyped and imputed SNPs was performed using UNPHASED (30), with two ancestry principal components as covariates and including only genotypes imputed with probability >0.9 in the analysis.
We tested specifically for association with a set of 84 candidate genes previously implicated in susceptibility to depression through their function or previous genetic association studies (see Supplementary Table 2 for list of genes, which was selected prior to genome-wide analysis). Results from the logistic regression analysis for 2,654 SNPs lying within the genes and in 20-kb flanking regions were extracted.
Meta-analysis was then performed on the U.K., Munich, and Lausanne studies. For the Munich and Lausanne studies, genome-wide summary statistics on the autosomal chromosomes were available (13). The Munich study had been genotyped on the Illumina 550K array, which overlaps substantially with the Illumina 610K array used in the U.K. study. For the Lausanne study, results from the same set of SNPs were available, which had been imputed where necessary, from genotypes on Affymetrix 500K chip. Meta-analysis was performed using the software METAL (www.sph.umich.edu/csg/abecasis/metal/).
Results
Association Analysis
In the analysis testing for association with 1,636 depression cases and 1,594 screened comparison subjects at 471,747 SNPs that passed quality control procedures, no single SNP reached genome-wide significance. Four SNPs showed suggestive evidence for association: two SNPs in BICC1 on chromosome 10q21.1 (rs9416742 [p=1.3×10–7] and rs999845 [p=3.1×10–7]), rs2698195 on chromosome 2p15, which is located 2 kb 5′ of KIAA1841, and rs8050326 (16q24.1, located over 100 kb from IRF8). The genomic control parameter (γ) decreased from 1.04 in the Cochran Armitage Trend test to 1.02 after accounting for two ancestry principal components, indicating little difference between depression cases and comparison subjects due to population ancestry or other systematic genomic effects. Details of the 20 SNPs with strongest signals for association are given in Table 1, with p values also provided from the meta-analysis with Munich and Lausanne cohorts.
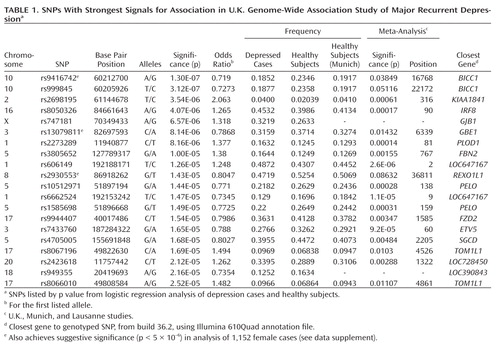 |
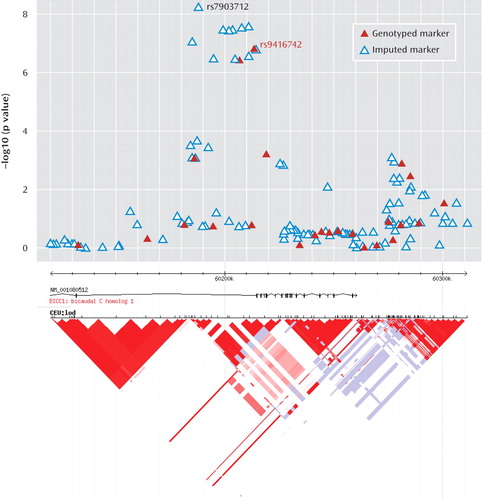
aStrongest evidence for association occurred at rs7903712 (p=5.7×10–9; imputation r2 =0.9685).
Analysis of imputed genotypes in the BICC1-region of 10q21 strengthened the evidence for association. Genome-wide significant evidence for association was detected at six SNPs in strong linkage disequilibrium, with the strongest evidence for association occurring at rs7903712 (Figure 1). At the genotyped SNP rs9416742, the minor allele, which had a frequency of 0.235 in healthy subjects, conferred a protective effect against depression (odds ratio=0.719, 95% CI=0.6362–0.8127). The SNPs rs9416742 and rs7903712 were regenotyped in-house (see supplementary materials) and showed strong concordance with Illumina 610Quad BeadChip genotyping (concordance 99.95% for rs9416742) and with imputed genotypes (98.37% for rs7903712). For both SNPs, mismatch was uncorrelated with genotype. Association testing with regenotyped data showed a similar pattern of results, with rs7903712 having higher significance levels than rs9416742 (p=9.65×10–8 versus p=3.07×10–6). The decrease in significance reflects the higher missing rates for genotypes (10.5% and 8.6%); these genotypes were not used in subsequent analysis.
 |
To explore the source of increased significance further, we analyzed haplotypes at these two markers. For rs7903712, imputed genotypes with estimated accuracy >90% were included, giving genotypes for 95.1% of the sample. Comparison with our regenotyped samples showed imputation missingness or genotype discordancy was not correlated with genotype. Only three haplotypes for rs9416742 and rs7903712 exist at frequency >1%. The increased evidence for association at rs7903712 was due to a haplotype of estimated frequency 1.7% in comparison subjects, which carried the common allele at rs9416742 but conferred a protective effect (Table 2). In HapMap, the additional protective haplotype identified by rs7903712 has a frequency of 2.5%.
Results for top 20 SNPs for sex-specific analyses are given in Supplementary Tables 3 and 4. In women (1,152 cases), genome-wide association was observed with rs9416742 in BICC1 (p=1.8×10–8; odds ratio=0.673, 95% CI=0.586–0.772), but men had a much weaker p value (p=0.039; odds ratio=0.830, 95% CI=0.695–0.991). A test for interaction between sex and genotype was not significant (p=0.149). In women, four further SNPs achieved suggestive significance. Three of these SNPs (rs8067196, rs2930553, and rs13079811) also showed strong association in the full study and are listed in Table 1. Suggestive evidence of association was also found at SNP rs987390, which lies 180 kb from rs2930553, near the REXO1L1 gene, strengthening association in this genetic region. In men (N=477), only a single SNP, rs6989226, near the TUSC3 gene reaches suggestive significance (p=1.81×10–6). No additional significant findings were identified using other genetic models (genotypic, dominant, and recessive).
In the analysis of 84 candidate genes, the strongest evidence for association was at an intronic SNP, rs13050655, in PDE9A (p=3.58×10–5, ranked 27 in the genome-wide analysis), a gene postulated to contribute to vulnerability to depression (31). Results by gene are shown in Supplementary Table 2.
Meta-Analysis
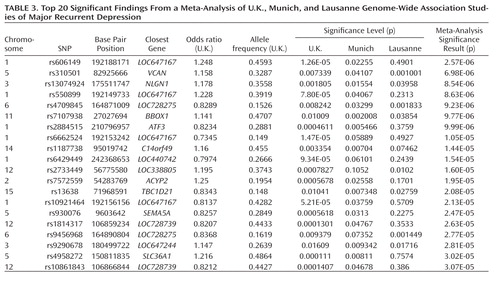 |
In the meta-analysis of U.K., Munich, and Lausanne studies, no SNP attained genome-wide significance and one SNP, rs606149, achieved suggestive significance (p=2.57×10–6). This SNP lies close to two other SNPs with p values of <2×10–5 in the analysis of U.K. cases and healthy comparison subjects (Table 1). The SNPs are located near LOC647167 on chromosome 1, which is otherwise a gene desert. The top 20 significant findings are shown in Table 3. Notably, the third strongest finding is rs13074924, which is located 29.7 kb from the 3′ end of NLGN1 (neuroligin 1) in 3q26.31, and >100 kb from the next gene. This SNP achieves modest p values in all three studies (p=0.001805 in the U.K. study, p=0.01554 in the Munich study, and p=0.03958 in the Lausanne study) with a consistent direction for allele effect, giving p=8.54×10–6 in the meta-analysis. In the meta-analysis, no replication was seen for the association with BICC1 in the U.K. study. In the Munich, Germany, and Lausanne cohorts, rs9416742 had p values of 0.11 and 0.06, respectively, but in the opposite direction to the effect in the U.K. cohorts. Of the SNPs reported in the U.K. study (Table 1), only those near LOC647167 have a p value of <1×10–4 in the meta-analysis.
Discussion
This genome-wide association study has found evidence for a role of genetic variants in the bicaudal C homologue 1 gene (BICC1) in recurrent unipolar major depression, however the finding that a minor allele of these variants has a protective effect against depression was not replicated in two independent samples of recurrent major depression collected and assessed with comparable procedures. The association of BICC1 and depression is a novel finding, with no previous published evidence for a role of this gene in neuropsychiatric disease.
The BICC1 gene product is an RNA-binding protein which has been found to form complex interactions with RNA and with other proteins. Several independent lines of evidence suggest a role for BICC1 in neurogenesis and plasticity, defects which may predispose to mood disorders. In Drosophila, it is required for dorsoventral patterning of the oocyte (32). The RNA binding activity of the protein is mediated by its KH domains (see Supplementary Figure 5) (33), which are characteristic of the heterogeneous nuclear ribonucleoprotein k family of transcription factors and bind both DNA and RNA (34). Other heterogeneous nuclear ribonucleoproteins are known to bind targets such as the serotonin transporter gene promoter (35). In humans, the C-terminus of the longer isoform-1 of the protein contains a sterile alpha motif (SAM) domain (Supplementary Figure 5), thought to be a highly specific postsynaptic targeting signal (36). This is interesting as our observed association is intronic to the long isoform, but is 15 kb 5′ from the first reported exon of a shorter isoform that does not contain a SAM domain and may highlight a haplotype which favors the expression of one or other of these isoforms. Both BICC1 isoform RNAs are expressed widely in normal tissues and are expressed in all brain regions including cerebral cortex, hippocampus, and midbrain (see Supplementary Figures 6a-d), although BICC1 isoform-1 RNA is more highly expressed than isoform-2, particularly in nerve tissue. BICC1 can uncouple dishevelled-2 signaling from the canonical Wnt pathway in a SAM domain dependent manner, suggesting that the different BICC1 isoforms may play different biological roles (37). Gene expression studies (Array Express; http://www.ebi.ac.uk/microarray-as/ae/) suggest that BICC1 is also upregulated by sodium valproate (a mood stabilizer and Wnt signaling inhibitor used to treat bipolar disorder), with approximately twofold increase in BICC1 expression after valproate treatment.
The other notable result is the meta-analysis finding of association with a SNP near neuroligin-1 (NLGN1) on 3q26.31. Echoing our BICC1 finding, it is known that all NLGNs are enriched at postsynaptic densities (38). NLGN1 encodes a neuronal cell surface protein and a splice site-specific ligand for beta-neurexins. The Ca(2+)-dependent neurexin/neuroligin complex is required for efficient neurotransmission, and involved in the formation of synaptic contacts. Neuroligins are likely to be part of the machinery employed during the formation and remodeling of CNS synapses. The neuroligin/neurexin complex has also been implicated in schizophrenia (39), with deletions of NLGN1 observed in autism (40); deletions of the closely related NLGN3 and NLGN4/5 result in a variety of psychiatric phenotypes (41, 42).
Several aspects of the finding in BICC1 may be relevant to replication and follow-up studies. First, the strongest association was found with a SNP that was not genotyped on the Illumina panel but could be imputed with high accuracy based on genotyped markers in strong linkage disequilibrium. The high accuracy of imputation was confirmed by targeted regenotyping, and the association was statistically significant after accounting for uncertainty of imputation and for potential multiple testing of all common variants across the human genome (27, 28). However, this effect did not replicate in either the Munich or Lausanne samples. Such nonreplication occurs frequently; with the modest effect sizes expected for genetic contributions to depression, associations will remain elusive with large variation across studies. Even for a true association, the effect size may be inflated in the discovery sample (43, 44).
Replication of the BICC1 finding will be necessary to determine whether this is a false positive result or a true effect which is also detectable in other studies. Such analyses are ongoing in the Psychiatric GWAS Consortium (45), of which this study is a part. The large, well-characterized samples that form the Psychiatric GWAS Consortium will be required before we can expect to see consistent signals of association through meta- and mega-analysis. The Psychiatric GWAS Consortium analysis should help resolve the role of BICC1 and other putative associations in depression. However, replication is likely to remain a substantial challenge for all psychiatric disorders. While large sample sizes increase the power to detect genetic risk loci, variation between studies will be unavoidable, and potentially unquantifiable. Sources of diversity between studies include those related to phenotype (e.g., criteria for depression, assessment measures of depression for both cases and comparison subjects), those related to genotype (e.g., population ancestry, genotyping panel used), and more subtle effects such as differences in patient ascertainment methods by study or cultural differences in seeking help for depression, which may result in heterogeneous case samples even though all patients meet clinical criteria for recurrent depression.
In order to evaluate the present results and to make a qualified comparison with other studies, the strengths and limitations of the study should be considered. The study used recurrent major depression, the type of depression that has been shown to be most heritable (4, 8), with collection of cases in the U.K. by the same clinical team giving a homogeneous clinical sample. As depression is common in the general population, excluding cases of lifetime depression from the comparison sample can substantially increase power of a study. We have therefore used a sample of comparison subjects screened to exclude those with higher liability to depression. The ability of the present study to detect true associations was limited by its sample size, which was comparable to the previously published genome-wide association studies (12, 13).
In conclusion, the present study provides evidence for a gene encoding a Wnt signaling component, BICC1, in the pathogenesis of depression. The meta-analysis implicated NLGN1, whose role in formation and remodeling of CNS synapses makes it a convincing functional candidate gene for depression. However, replication of these findings in other studies will be essential to confirm their role in depression. The results of this and other genome-wide association studies in depression suggest that effects of common genetic variants on depression are individually weak and multiple large samples will be needed to provide a more comprehensive picture of common genetic variants in depression.
1 : Psychological autopsy studies of suicide: a systematic review. Psychol Med 2003; 33:395–405 Crossref, Medline, Google Scholar
2 ; DELTA Study Group: Health related quality of life in recurrent depression: a comparison with a general population sample. J Affect Disord 2010;120:126–132 Crossref, Medline, Google Scholar
3 : The global burden of disease, 1990–2020. Nat Med 1998; 4:1241–1243 Crossref, Medline, Google Scholar
4 : A hospital-based twin register of the heritability of DSM-IV unipolar depression. Arch Gen Psychiatry 1996; 53:129–136 Crossref, Medline, Google Scholar
5 : Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000; 157:1552–1562 Link, Google Scholar
6 : The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 2003; 60:497–502 Crossref, Medline, Google Scholar
7 : Polycystic ovarian syndrome in a woman with polycystic kidney disease. Eur J Obstet Gynecol Reprod Biol 2008; 140:282–283 Crossref, Medline, Google Scholar
8 : Reliability of a lifetime history of major depression: implications for heritability and co-morbidity. Psychol Med 1998; 28:857–870 Crossref, Medline, Google Scholar
9 : The lifetime history of major depression in women: reliability of diagnosis and heritability. Arch Gen Psychiatry 1993; 50:863–870 Crossref, Medline, Google Scholar
10 : Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry 2008; 13:772–785 Crossref, Medline, Google Scholar
11 : Homing in on depression genes. Am J Psychiatry 2007; 164:195–197 Link, Google Scholar
12 : Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry 2009; 14:359–375 Crossref, Medline, Google Scholar
13 : Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry (advance online publication, December 23, 2008;
14 : Depression Case Control (DeCC) Study fails to support involvement of the muscarinic acetylcholine receptor M2 (CHRM2) gene in recurrent major depressive disorder. Hum Mol Genet 2009; 18:1504–1509 Crossref, Medline, Google Scholar
15 : The Depression Network (DeNT) Study: methodology and sociodemographic characteristics of the first 470 affected sibling pairs from a large multi-site linkage genetic study. BMC Psychiatry 2004; 4:42 Crossref, Medline, Google Scholar
16 : Whole genome linkage scan of recurrent depressive disorder from the depression network study. Hum Mol Genet 2005; 14: 3337–3345 Crossref, Medline, Google Scholar
17 : Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J 2009; 9:225–233 Crossref, Medline, Google Scholar
18 : Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259 Crossref, Medline, Google Scholar
19 : SCAN: Schedules for Clinical Assessment in Neuropsychiatry. Arch Gen Psychiatry 1990; 47:589–593 Crossref, Medline, Google Scholar
20 : GENESiS: creating a composite index of the vulnerability to anxiety and depression in a community-based sample of siblings. Twin Res 2000; 3:316–322 Medline, Google Scholar
21 : Past and present state examination: the assessment of "lifetime ever" psychopathology. Psychol Med 1986; 16:461–465 Crossref, Medline, Google Scholar
22 : Family history of depression is associated with younger age of onset in patients with recurrent depression. Psychol Med 2008; 38:641–649 Crossref, Medline, Google Scholar
23 : The PsyCoLaus study: methodology and characteristics of the sample of a population-based survey on psychiatric disorders and their association with genetic and cardiovascular risk factors. BMC Psychiatry 2009; 9:9 Crossref, Medline, Google Scholar
24 : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38:904–909 Crossref, Medline, Google Scholar
25 : Genomic control for association studies. Biometrics 1999; 55:997–1004 Crossref, Medline, Google Scholar
26 : PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–575 Crossref, Medline, Google Scholar
27 : Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 2008; 32:227–234 Crossref, Medline, Google Scholar
28 : Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008; 32:381–385 Crossref, Medline, Google Scholar
29 : Mach 1.0: rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet 2006; S79:2290 Google Scholar
30 : Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered 2008; 66:87–98 Crossref, Medline, Google Scholar
31 : Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proc Natl Acad Sci U S A 2006; 103:15124–15129 Crossref, Medline, Google Scholar
32 : Bicaudal C and trailer hitch have similar roles in gurken mRNA localization and cytoskeletal organization. Dev Biol 2009; 328:434–444 Crossref, Medline, Google Scholar
33 : K homology domains of the mouse polycystic kidney disease-related protein, bicaudal-C (Bicc1), mediate RNA binding in vitro. Nephron Exp Nephrol 2008; 108:e27–e34 Crossref, Medline, Google Scholar
34 : Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J Biol Chem 1995; 270:4875–4881 Crossref, Medline, Google Scholar
35 : YB-1 and CTCF differentially regulate the 5-HTT polymorphic intron 2 enhancer which predisposes to a variety of neurological disorders. J Neurosci 2004; 24:5966–5973 Crossref, Medline, Google Scholar
36 : Efficient targeting of proteins to post-synaptic densities of excitatory synapses using a novel pSDTarget vector system. J Neurosci Methods 2009; 181:227–234 Crossref, Medline, Google Scholar
37 : Bicaudal C, a novel regulator of Dvl signaling abutting RNA-processing bodies, controls cilia orientation and leftward flow. Development 2009; 136:3019–3030 Crossref, Medline, Google Scholar
38 : Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008; 455: 903–911 Crossref, Medline, Google Scholar
39 : Dual constraints on synapse formation and regression in schizophrenia: neuregulin, neuroligin, dysbindin, DISC1, MuSK and agrin. Aust N Z J Psychiatry 2008; 42:662–677 Crossref, Medline, Google Scholar
40 : Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459:569–573 Crossref, Medline, Google Scholar
41 : A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007; 318:71–76 Crossref, Medline, Google Scholar
42 : Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet 2008; 16:614–618 Crossref, Medline, Google Scholar
43 : Large upward bias in estimation of locus-specific effects from genomewide scans. Am J Hum Genet 2001; 69:1357–1369 Crossref, Medline, Google Scholar
44 : Validating, augmenting and refining genome-wide association signals. Nat Rev Genet 2009; 10:318–329 Crossref, Medline, Google Scholar
45 : Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009; 166:540–556 Link, Google Scholar

