Huntington’s Disease: Getting Closer
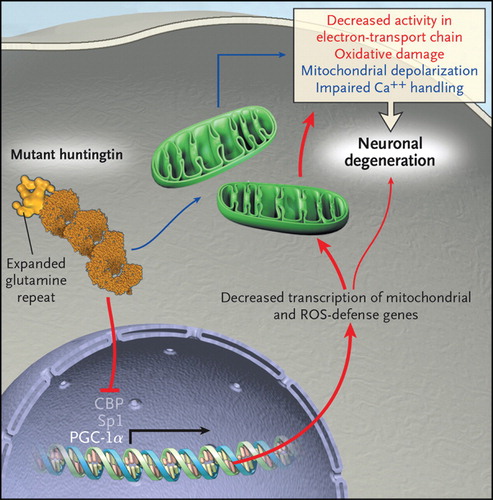
Huntington’s disease runs in families, and it devastates them with its relentless and painfully predictable assault on what makes us human: our motor, cognitive, and affective functions. Astonishingly, almost 15 years after the discovery of the causative gene mutation, our understanding of disease pathogenesis is still rudimentary. We know that the responsible gene is located on the short arm of chromosome 4, and we know that it contains a variable stretch of repeating CAG trinucleotides, or codons, each of which codes for a glutamine residue in the huntingtin protein. Individuals with 40 or more repeats will develop Huntington’s disease; those with 35 or fewer will not. But how this polyglutamine expansion in huntingtin causes the disease is uncertain. Is it due, in part, to a loss of normal huntingtin function, whatever that is? Or is it caused primarily by the ability of the mutant protein to gain a new and toxic function? Fifteen years out, these remain fundamental questions, and the therapeutic implications will be quite different, depending on the answers. Hypotheses of Huntington’s disease pathogenesis have come and gone, but a few have had staying power, including that mutant huntingtin 1) alters gene transcription and thereby changes the set of proteins expressed by affected cells; 2) directly affects mitochondria, reducing their ability to produce energy and to buffer calcium, and indirectly increases their production of reactive oxygen species (ROS); or 3) reduces the activity of peroxisome proliferator-activated receptor-g coactivator 1a (PGC-1a), a transcriptional coactivator that regulates many metabolic processes, including mitochondrial genesis and antioxidant defenses. There is great interest in the possibility that these putative pathogenic processes are interrelated, as depicted in the figure , but this is undoubtedly an oversimplification. Nevertheless, this formulation suggests testable hypotheses and novel therapeutic strategies. Huntington’s disease will continue to run in families for the foreseeable future, but perhaps we are on the verge of lessening its devastation.