
Evidence for Onset of Antipsychotic Effects Within the First 24 Hours of Treatment
Abstract
OBJECTIVE: It is widely held that there is a delayed onset of antipsychotic action and that any early effects represent nonspecific behavioral effects. Recent research has shown that antipsychotic action begins within the first week. The authors tested the hypothesis that psychosis improves within the first 24 hours of antipsychotic treatment. METHOD: In this multicenter, double-blind, placebo-controlled study, 311 patients with a diagnosis of schizophrenia spectrum disorder and an acute exacerbation were randomly assigned to receive 10 mg i.m. of olanzapine, 7.5 mg i.m. of haloperidol, or intramuscular placebo. Subjects were rated with structured rating scales (Positive and Negative Syndrome Scale and Clinical Global Impression) at baseline, 2 hours, and 24 hours. RESULTS: The olanzapine and haloperidol groups showed greater resolution of overall symptoms than the placebo group; for the olanzapine group, this effect was evident at 2 hours. A factor analysis showed that an independent change in psychosis (which included conceptual disorganization, hallucinatory behavior, unusual thought content) was evident within the first 24 hours for both drugs. This improvement in core psychosis was not mediated unidirectionally by changes in nonspecific behavioral effects or other psychopathology. CONCLUSIONS: These data suggest that the onset of antipsychotic action is early and that the magnitude of this action grows with time. This clinical reality calls into question some prevailing hypotheses regarding the mechanism of action of antipsychotics and suggests that antipsychotic action may be more proximally related to the blockade of dopamine transmission than was originally thought.
Antipsychotics were introduced to modern psychiatric treatment more than half a century ago, yet their mechanism of action remains an issue of active debate (1). A central question regarding antipsychotic action is the speed of “onset” of antipsychotic response. It is widely held that there is a “delayed onset” of antipsychotic response, somewhere in the range of 2–3 weeks, an idea now embedded in standard psychiatric texts (2, 3). The earlier effects are thought to be a reflection of the nonspecific behavioral effects of antipsychotics on aspects such as agitation, excitement, and uncooperativeness. However a recent meta-analysis by Agid et al. (4), which included data from 7,450 patients in 42 double-blind, active-drug or placebo-controlled trials, did not support this “delayed onset” hypothesis. The authors found that 1) there is a definite change in psychosis by the end of the first week, 2) the degree of improvement in the first week exceeds that in each subsequent week, and 3) this improvement is over and above any change accorded to placebo or nonspecific behavioral improvement (4). If antipsychotic response is established within the first week, how early can it be apparent?
When antipsychotics were introduced in the 1950s and 1960s, it was routinely noted that they were effective within the first few days (5, 6). In the 1970s the interest in “rapid neuroleptization” (7) led to several studies that compared the safety and efficacy of different doses, parenteral versus oral administration, and low-potency versus high-potency agents in the first few days of treatment (8). However, 1) many of these reports included small and uncontrolled case series (7); 2) they did not include a placebo control (7, 9–15); 3) raters were not blind to treatment assignment (7, 14); 4) the response measures reported often did not differentiate between effects on psychosis and nonspecific effects on behavior (10, 13); and 5) where the two measures were separately reported, both were shown to improve, and none of these studies tested whether the improvement in psychosis is distinct from, or secondary to, a nonspecific behavioral effect (9, 11, 12, 14–16). These studies all pointed to a rapid response—however, they suffered from two major confounding factors. First, most of these studies lacked a placebo control. The patients included in studies of acute exacerbation were often at the apogee of their worsening, and investigators could have had a bias to overestimate this worsening to include them in the trial. Both of these factors may lead to the appearance of a rapid response—an issue that can only be resolved with the inclusion of a placebo-treated control group. Second, these studies did not include appropriate subscales or did not carry out the required analyses that permit one to determine whether this early response in psychosis is primary or secondary to the effects of the drug on agitation and excitement.
A very good opportunity to test this question was provided by a recent large-scale clinical trial that compared the efficacy of haloperidol, olanzapine, and placebo in patients with schizophrenia during the first 24 hours of treatment (17). Unlike the previous reports, this study 1) was large (N=311), 2) was placebo-controlled, 3) had raters who were blind to drug assignment, 4) used standardized and comprehensive rating scales (Positive and Negative Syndrome Scale and Brief Psychiatric Rating Scale [BPRS]) with separable items for psychosis and agitation, and 5) obtained data at 2 hours and then at 24 hours after the onset of treatment. Using these data we investigated the following questions: Is there an improvement in core psychotic symptoms in the first 24 hours? Is this effect clearly different from the effect of placebo? Is this early improvement in psychosis a secondary consequence of nonspecific tranquilizing effect of the drugs?
Method
Study Subjects
The study was an Eli Lilly–sponsored, multisite, international study comparing two active drugs (haloperidol and olanzapine) in their intramuscular form versus placebo in a double-blind, randomized, controlled trial. Local ethics review boards approved the study protocol, including the use of placebo, given the hospitalized status of all participating patients, the 2:2:1 randomization ratio for active treatment versus placebo, the brief duration of the study (24 hours), and the use of active medication based on the clinical judgment of the investigator at the time of randomization. Written informed consent was obtained from all patients and from a relative or legal representative when required by local law or custom.
Recently hospitalized patients ages 18 years or older who had been assessed by the study investigators and given a clinical diagnosis of DSM-IV schizophrenia, schizophreniform disorder, or schizoaffective disorder and who exhibited an episode of acute agitation in the context of psychosis were included. Patients were included in the study if they demonstrated a total score of 14 or higher (of a maximum of 35) on the Positive and Negative Syndrome Scale excitement component (which included items measuring tension, uncooperativeness, hostility, poor impulse control, and excitement) with at least one item score >4. Patients with significant, unstable, medical disorders and those who were too agitated to provide informed consent or to cooperate with the requirements of the study were not included in this trial.
Clinical Design and Outcome Measures
The study consisted of a screening period and a 24-hour intramuscular treatment period. Patients were not allowed to receive any antipsychotic treatment during the screening period, which lasted for a minimum of 2 hours. On entering the treatment period, patients were randomly allocated to treatment with 10 mg i.m. of olanzapine, 7.5 mg i.m. of haloperidol, or intramuscular placebo in a 2:2:1 patient ratio. The dose of intramuscular olanzapine was based on previous open-label clinical trials, and the dose of intramuscular haloperidol was chosen after a literature review and discussions with ethics boards, regulatory authorities, and participating psychiatrists. Both drugs and placebo were administered in identical, color-blinded, translucent standard syringes. Raters and study personnel were blind to treatment assignment. Optional second and third injections were given 2 or more and 4 or more hours following the first and optional second injections, respectively. Other medications affecting the central nervous system and prophylactic anticholinergics were prohibited. Benzodiazepines were allowed at the discretion of the treating clinician, but only for patients who received more than one injection of active treatment/placebo. Those who received two injections of drug/placebo could receive a dose of a benzodiazepine (2 mg of lorazepam or equivalent) no earlier than 1 hour after the injection. Those who received a third injection of the drug/placebo could get the second dose of benzodiazepine at least 1 hour after that injection.
The study investigators assessed patients at the screening visit and immediately before and at 2 and 24 hours after the first injection. A full 18-item BPRS scale plus one item derived from the Positive and Negative Syndrome Scale (“poor impulse control”) was administered at baseline and at 2 and 24 hours. The BPRS items were rated by using the Positive and Negative Syndrome Scale anchors, as this approach provided additional reliability. Data from the “poor impulse control” item were collected because this item, along with four other BPRS items (tension, uncooperativeness, hostility, and excitement), constituted a validated and independent factor for assessing agitation and excitement, termed the Positive and Negative Syndrome Scale excitement component (18). Additional details of the study and an analysis focusing on the outcome of agitation as measured by the excitement component of the Positive and Negative Syndrome Scale were published earlier (17).
Approach to Analysis
The main intent of this study was to determine whether there was drug-induced early (within first 24 hours) improvement in psychosis and how this effect was related to drug-induced changes in nonspecific behavioral symptoms. Although the BPRS scale has been subjected to factor analysis, most of these analyses were derived from large-scale studies of stable patients, and it is now clear that the factor structure is not invariant over time (19). Because our primary interest was in the pattern of change in the first 24 hours, we undertook a factor analysis of the change scores on all the rated items of the BPRS to get an empirically valid estimate of the factors of early antipsychotic response. To relate this factor-analysis-derived answer to more conventional measures, we also asked whether the improvement in the conventional BPRS factor representing psychosis (items: conceptual disorganization, hallucinatory behavior, grandiosity, unusual thought content) was explained by changes in the conventional BPRS factors representing activation and hostility (items: excitement, tension, mannerisms, suspiciousness, hostility, uncooperativeness). Further, because the five items that constitute the Positive and Negative Syndrome Scale excitement component are also markers of nonspecific behavioral effects and have shown to improve with drug treatment in 24 hours, we examined if this factor could explain change in the BPRS psychosis factor. Finally, we used the mediator analysis strategy of Kraemer et al. (20) to examine if these early changes in core psychosis could be explained by changes in any of the nonspecific factors. Statistical testing, as described in the following sections, was performed with the SAS System version 8.2 (SAS, Cary, N.C.). An alpha of 0.05 for significance was used.
Determining the Factors of Early Response
The percentage of reduction in baseline score was calculated for each Positive and Negative Syndrome Scale–derived BPRS item at 2 hours and 24 hours. The following equation was used to calculate these changes:
where yij is the change in baseline score for BPRS item i at time j; xij is the raw score for BPRS item i at time j; xi0 is the baseline score for BPRS item i; i is the BPRS item, ranging from 1–18; and j is the time since onset of study, equal to 2 hours or 24 hours.
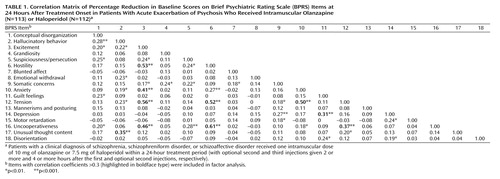
To understand the nature of the relationship between these changes at a given point in time, we developed a correlation matrix of the 24-hour change scores (Table 1), and the data were subjected to a series of factor analyses. Because we were interested in identifying factors in drug-induced olanzapine or haloperidol change at this stage, the first factor analyses were based on the data from patients who received active treatment. Subsequent comparisons included the data from patients who received placebo.
An unweighted least-squares approach was implemented, and factor solutions were then rotated using a varimax (orthogonal) rotation. Initially, all 18 items were incorporated into the analysis, and those items that did not contribute meaningfully to any of the factors (i.e., had a maximal loading less than 0.30) were removed from the analysis. This step was done to conserve statistical power and to make the analysis and its interpretation as clean and simple as possible.
Drugs Versus Placebo Effects on Factor Scores
Factor scores were calculated for each patient by adding up their change scores for each of the items that loaded on a corresponding factor. Some items loaded into more than one factor (e.g., excitement loaded on both factor 1 and factor 2), in which case they were added to both factor scores. Differences in the degree of improvement in these factors with drug versus placebo were compared at the 2 and 24 hour time points by using analysis of variance. If the overall model was significant, post hoc t tests were done to ascertain the statistical significance of differences at given time points. A Bonferroni adjustment, relative to the number of multiple comparisons in each model, was applied to the p values as a conservative way to reduce the risk of finding false positive results and to maintain an overall alpha level of 0.05.
Changes in Other Factors
To test whether changes in other factors accounted for the improvement in psychosis, a series of mediator analyses and analyses of covariance (ANCOVAs) was performed. The definitions of mediator/moderator used in these analyses are those defined by Kraemer et al. (20). By definition, change scores cannot be moderators because they are not baseline characteristics that occur independent of and prior to treatment. Thus, we were left with only the question of whether any of the factor scores were mediating the other factors.
The method proposed by Kraemer et al. (20) involves the construction of three equations:
| 1. | mediator=β0 + β1 treatment | ||||
| 2. | outcome=β2 + β3 treatment | ||||
| 3. | outcome=β4 + β5 mediator + β6 treatment + β7 mediator × treatment | ||||
where β0, β2, and β4 are intercept terms; β1, β3, and β6 are coefficients associated with the magnitude of the treatment effect; β5 is a coefficient associated with the magnitude of the mediator effect; and β7 is a coefficient associated with the magnitude of the mediator/treatment interaction.
To test the hypothesis that change in psychosis is secondary to improvements in other aspects measured by the BPRS scale, we tested the hypothesis of whether the other factors were “mediators” of the change in core psychosis. In accordance with the equations, the factor representing core psychosis was treated as the “outcome” and others were treated as “mediators.” To see if the mediation was bidirectional, the other factors were treated as “outcome” and the psychosis factor was treated as a “mediator.” Mediation is said to occur when all of the following conditions are met:
| 1. | The mediator is correlated with treatment. | ||||
| 2. | The effect of treatment (β1) is significant in equation 1. | ||||
| 3. | The effect of treatment (β3) is significant in equation 2. | ||||
| 4. | The mediator has a significant effect (β5) in equation 3. | ||||
In cases where bidirectional mediation was found to occur, tests were performed to determine whether mediation was occurring more strongly in one direction than in the other. In these tests, the mediator and the outcome measure were both standardized, and then the coefficients associated with the mediator in equation 3 were compared across the two directions. A 95% confidence interval for each of these coefficients was constructed, and mediation was considered to occur equally in both directions if these two intervals overlapped. If they did not overlap, the dominant direction of the relationship could then be determined.
As another convergent test of the hypothesis, we addressed the issue of whether treatment has any effect on change in psychosis above and beyond reduction in nonspecific behavioral factors. A series of ANCOVAs (analysis of covariance) was performed to address this issue, in which measures of change in psychosis were used as the outcome variables, treatment was included as an explanatory variable, and change in nonspecific behavioral factors was included as a covariate. If the effect of treatment was found to be significant after adjustment for the relationship between the outcome measure and the covariate, then we could conclude that treatment had an additional effect on psychosis beyond merely reducing nonspecific behavioral factors.
Results
A total of 311 patients were included in the study and were randomly assigned to receive intramuscular olanzapine (N=131), intramuscular haloperidol (N=126), or intramuscular placebo (N=54). Baseline and 2-hour data were available for 286 (92%) patients, and data to assess changes at 24 hours were available for 273 (88%) patients, including 113 patients who received olanzapine, 112 who received haloperidol, and 48 who received placebo. There were no significant between-group baseline demographic or illness differences. The mean age of the patients was 38.2 years (SD=11.6, range=18–72); their mean age at onset of illness was 24.4 (SD=8.5, range=7–58). Thirty-nine percent (N=21) of the placebo group required the use of adjunctive benzodiazepines during the first 24 hours, compared to only 15% of the active treatment group (21 patients who received haloperidol and 16 who received olanzapine). The difference between the placebo and active treatment groups was significant (p<0.0001, Fisher’s exact test). The majority of patients who received adjunctive benzodiazepines received only one dose, and although more patients in the placebo group received benzodiazepines, there was no difference in the number and timing of benzodiazepine administration across the groups (χ2=1.02, df=2, p=0.60).
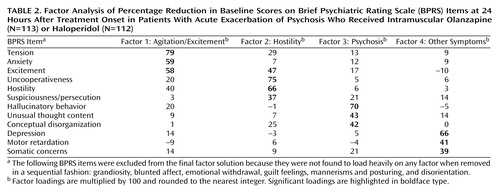
To determine the different dimensions of improvement (change in scores) after antipsychotic treatment, an unweighted least-squares approach was implemented, and three solutions, with three, four, and five factors, respectively, were obtained. Although all three solutions (Table 1) yielded similar findings, the four-factor solution was selected for further analysis for the following reasons: 1) all factors in this solution contained at least three significant item loadings, 2) this solution resulted in the least amount of overlap between the factors (i.e., the fewest cases where items loaded significantly on more than one factor), and 3) the items clustered within each factor could be interpreted in a clinically meaningful manner.
As Table 2 shows, factor analysis segregated items associated with agitation and excitement into the first factor (called agitation/excitement factor) and items associated with hostility into the second factor (hostility). The third factor contained items that measure psychosis (conceptual disorganization, hallucinatory behavior, and unusual thought content). The fourth factor brought in a few more distinct items (somatic concerns, depression, and motor retardation) that were not found to be significant in the three-factor solution. Relevant to our hypothesis, we found that change in agitation-related items segregated independent of improvement in psychosis-related items.
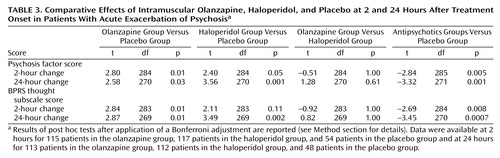
The analysis of factor scores revealed that both intramuscular olanzapine and intramuscular haloperidol show a significant effect on the psychosis factor at 24 hours, compared to placebo (Table 3).
In the case of intramuscular olanzapine, a significant effect on the psychosis factor was evident as early as 2 hours (t=2.80, df=284, p=0.01, with Bonferroni adjustment). Haloperidol was not differentiated from placebo at the 2-hour time point, although the difference approached significance (t=2.40, df=284, p=0.05, with Bonferroni adjustment). Intramuscular haloperidol and intramuscular olanzapine did not differ statistically in the degree of improvement on this factor at 2 hours or 24 hours.
The same question was also addressed by using the more conventional subscale that addresses core psychotic symptoms—the BPRS thought subscale. With that collection of items, intramuscular olanzapine was differentiated from placebo at 2 hours (t=2.84, df=283, p=0.01, with Bonferroni adjustment), and both intramuscular olanzapine and intramuscular haloperidol were differentiated from placebo at 24 hours (t=3.45, df=270, p=0.0007, with Bonferroni adjustment). To test if this improvement in the BPRS was secondary to improvement in nonspecific behavioral factors, we used covariance to adjust for the effects of improvement in nonspecific behavioral factors. There was a statistically significant effect of the active treatments on the BPRS thought subscale score after correction for the BPRS activation-hostility subscale score (overall model: F=14.54, df=3, 267, p<0.0001; independent effect of active treatment: t=2.21, df=267 p=0.03). Similarly, there was a statistically significant effect of the active treatments on BPRS thought subscale scores after correction for Positive and Negative Syndrome Scale excitement component scores (overall model: F=11.79, df=3, 268, p<0.0001; independent effect of active treatment: t=2.58, df=268, p=0.01).
The mediation analysis revealed that the factors did explain the variance in each other (which suggests that they bear some relation); however, the mediation coefficients revealed bidirectionality. The size of the coefficient signifying mediation (β5 in equation 3) was as large when one considered the mediation of psychosis by changes in agitation-excitement (mean=0.36, SE=0.08) (p<0.0001, linear regression) as it was when one considered the mediation of agitation-excitement by psychosis (mean=0.34, SE=0.09) (p<0.0001, linear regression), thus refuting the hypothesis of unidirectional mediation of change in one by the other.
As a further confirmation of the overall hypothesis, the 2-hour change in agitation-excitement did not predict the 24-hour change in the psychosis factor (t=1.60, df=216, p=0.11). On the other hand, change in psychosis at the 2-hour mark predicted change in psychosis at the 24-hour mark (t=5.23, df=216, p<0.0001), suggesting that the early response in psychosis is distinct from changes in agitation-excitement and is continuous over time.
Discussion
There are no studies that have systematically examined the issue of early onset of antipsychotic response; however, results from a number of case series and uncontrolled studies have hinted at a rapid response to antipsychotic medications. Our study confirmed and extended these findings by testing them in a double-blind, placebo-controlled design and by showing that there are definite changes in measures of the psychotic component very early in treatment and that these changes are distinguishable from any changes in agitation or other nonspecific aspects of the acute presentation.
Our study has several limitations that must be taken into account in the interpretation and generalization of these results. First, the study was limited to patients who were demonstrating agitation or otherwise needed acute intervention. Although the results clearly demonstrate an improvement in psychosis, there still remains the question of whether quiet psychotic patients would also show a similar pattern of response. Agid et al. (4) showed that an unagitated psychotic group demonstrated a clear antipsychotic response by the first week, but they did not have the data to address responses earlier than the first week. This question remains an important, and tractable, issue for future clinical trials.
Second, the patients in this study had limited periods of washout, largely reflecting a clinical reality that required urgent action. In this situation, there is the possibility that previous treatment may have “primed” the patients for response, bringing them close to the threshold of response, and thus, the rather immediate response observed here may not be seen if the patients were drug naive or drug free for long periods. Although this explanation is possible, we believe it is unlikely, because almost all the previous studies (although not very well controlled) on this issue included drug-free or drug-naive patients and uniformly observed a rapid response (7, 9–16).
Third, the active treatment groups were less likely than the placebo group (15% versus 39%) to require the use of adjunctive benzodiazepines, as would be expected clinically. This difference works in favor of our hypothesis. That the antipsychotics were differentiated from placebo, despite twice as much use of adjunctive benzodiazepines (which provide sedation and behavioral control) in the placebo group further strengthens our claim of a specific and early antipsychotic action.
Fourth, the mode of administration of the antipsychotics in this study was intramuscular, which raises the question of whether a similar speed of onset of antipsychotic action would be observed had these medications been given orally. The intramuscular preparations certainly reach peak concentration earlier and have a higher level of bioavailability in the first few hours (21). For example, intramuscular olanzapine has a time to maximum plasma levels (Tmax) of 0.5 hours, while oral olanzapine has a Tmax of 3.5 hours (22); intramuscular haloperidol has a Tmax of 40–60 minutes (23), while the Tmax for oral haloperidol is in the range of 4–6 hours (24). The difference is only a matter of a few hours after the first dose, and overall systemic exposure after a given dose is nearly the same (22, 25). Furthermore, a single oral dose of 15 mg of olanzapine showed 79%–80% dopamine D2 receptor occupancy in the striatal and extrastriatal regions within 6 hours (26); a single oral dose of 4–7.5 mg of haloperidol showed greater than 80% occupancy of dopamine D2 receptors within 3 hours (27). Thus, while drug availability after administration of intramuscular agents may be somewhat faster (21), we expect that a similar pattern is likely to be observed with oral administration of an appropriate dose.
Finally, the study examined only haloperidol, a typical antipsychotic, and olanzapine, an atypical antipsychotic, and therefore the extension of these findings to other agents is not warranted. However, if the meta-analysis of Agid et al. (4) is any guide, this phenomenon is likely to be relevant to other antipsychotics as well.
The study findings raise some interesting implications from a clinical as well as a theoretical perspective. Clinicians have always known about the acute effects of antipsychotic medications—that is why they are a staple in every emergency department. However, it is largely assumed that these acute effects are nonspecific or “behavioral” and that the antipsychotic effects do not occur until much later. The current data suggest a reconsideration of this viewpoint. Not only are these early effects apparent on the psychosis dimension, the magnitude of these early effects is not trivial. The degree of improvement reported in the first few days is actually rather striking. For example, in the study by Stern et al. (14), nearly 50% of the improvement seen at the end of 2 weeks was evident by the 3-day mark. The study by Glovinsky et al. (16) showed a significant change within the first 3 days of treatment and a relatively limited additional improvement in the weeks that followed. The meta-analysis by Agid et al. (4) showed that more improvement in psychosis was observed in the first week than in any week thereafter. The present study had an extension component wherein patients’ medication was switched to the respective oral preparation after the first day (28). It is interesting to note that although differentiation from baseline occurred with both active drugs within the first day, with olanzapine’s effects seen within the first 2 hours, there was no further significant improvement observed between days 1 and 5 of treatment in the olanzapine and haloperidol groups (28). Given this finding, we think that the early response is not just statistically significant but clinically relevant as well.
This study raises further questions about the hypothesis of delayed onset of antipsychotic action and leads us to propose an “early-onset hypothesis.” It is well recognized that substantial blockade of the dopamine system by antipsychotic medications happens within the first few hours of treatment (26, 27). Because it was assumed all along that clinical response was delayed in onset, this dissociation between early occupancy and delayed onset of antipsychotic action was seen as a paradox. To explain this paradox, several explanations have been proposed. A widely considered explanation was that of a depolarization blockade. According to that hypothesis, the acute blockade of dopamine receptors led to an increased firing of dopamine neurons, which finally resulted in an exhaustion and depolarization blockade of the neurons 2–3 weeks down the line (29–31). This terminal event was seen as the mediator responsible for the delayed onset of antipsychotic response (31). Along slightly different lines, it has been proposed that the dopamine blockade leads to changes in gene expression, which over the ensuing days and weeks, lead to synaptic plastic changes, which are then seen as the proximal mediators of the onset of antipsychotic response (32). Although the depolarization blockade and synaptic plastic changes remain important empirical phenomena and may have a role in the longer-term consequences of antipsychotics, in light of the fact that the onset of antipsychotic response precedes these changes, their role as critical mediators of antipsychotic response may need to be reconsidered.
The realization of the early onset of antipsychotic response calls for a different kind of explanation than that suggested by the mechanisms proposed previously. A study by Abi-Dargham et al. (33) may provide an interesting clue in this regard. Using alpha-methyl-para-tyrosine, an agent that causes acute dopamine depletion (a fact verified by brain imaging in that study), they showed a robust antipsychotic response within the first 48 hours. Thus, the interruption of dopamine transmission at D2 receptors, whether by blockade or depletion, may be the immediate mediator of antipsychotic response (1). If that is the case, theories that relate alterations in dopamine transmission to their proximate effects on reward, motivational salience, and prediction error (34–38) may have more bearing on antipsychotic effect than accounts that focus on mechanisms that require 2–3 weeks for onset.
In conclusion, the study demonstrated that in acutely agitated psychotic patients with a schizophrenia spectrum disorder, the acute administration of a typical or atypical antipsychotic leads to a robust and independent improvement in psychotic symptoms. This improvement is observable as early as 2 hours after treatment with olanzapine and definitely by 24 hours with both olanzapine and haloperidol. This improvement is distinct from a placebo response and is not secondary to drug-induced changes in anxiety, agitation, or other nonspecific factors. These findings question the well-accepted “delayed-onset” hypothesis of antipsychotic action and call for a reconsideration of some of the clinical practices and scientific theories that have come in its wake.
 |
 |
 |
Received Jan. 13, 2004; revision received April 9, 2004; accepted May 10, 2004. From the Centre for Addiction and Mental Health; the Department of Psychiatry, University of Toronto, Toronto; Eli Lilly and Co., Indianapolis; and Eli Lilly Canada, Toronto. Address correspondence and reprint requests to Dr. Kapur, Centre for Addiction and Mental Health, 250 College St., Toronto, Ont., Canada M5R 1T8; [email protected] (e-mail). The data used in this analysis were provided by Eli Lilly and Co. No financial support was sought or received from Eli Lilly for this work. Dr. Lindborg and Dr. Jones are employees of Eli Lilly. Dr. Kapur’s research in this area is supported by a Canada Research Chair.
1. Kapur S, Remington G: Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry 2001; 50:873–883Crossref, Medline, Google Scholar
2. Gelder MG, Lopez-Ibor JJ, Andreasen NC: New Oxford Textbook of Psychiatry. New York, Oxford University Press, 2000Google Scholar
3. Sadock BJ, Sadock VA (eds): Kaplan and Sadock’s Comprehensive Textbook of Psychiatry, 7th ed. Philadelphia, Lippincott Williams & Wilkins, 2000Google Scholar
4. Agid O, Kapur S, Arenovich T, Zipursky RB: Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 2003; 60:1228–1235Crossref, Medline, Google Scholar
5. Casey J, Bennet I, Lindley C, Hollister L, Gordon M, Springer N: Drug therapy in schizophrenia: a controlled study of the relative effectiveness of chlorpromazine, promazine, phenobarbital and placebo. Arch Gen Psychiatry 1960; 2:210–220Crossref, Google Scholar
6. Guttmacher MS (National Institute of Mental Health Psychopharmacology Service center Collaborative Study Group): Phenothiazine treatment in acute schizophrenia: effectiveness. Arch Gen Psychiatry 1964; 10:246–261Crossref, Medline, Google Scholar
7. Donlon PT, Tupin JP: Rapid “digitalization” of decompensated schizophrenic patients with antipsychotic agents. Am J Psychiatry 1974; 131:310–312Link, Google Scholar
8. Donlon PT, Hopkin J, Tupin JP: Overview: efficacy and safety of the rapid neuroleptization method with injectable haloperidol. Am J Psychiatry 1979; 136:273–278Link, Google Scholar
9. Paprocki J, Versiani M: A double-blind comparison between loxapine and haloperidol by parenteral route in acute schizophrenia. Curr Ther Res Clin Exp 1977; 21:80–100Medline, Google Scholar
10. Tuason VB: A comparison of parenteral loxapine and haloperidol in hostile and aggressive acutely schizophrenic patients. J Clin Psychiatry 1986; 47:126–129Medline, Google Scholar
11. Neborsky R, Janowsky D, Munson E, Depry D: Rapid treatment of acute psychotic symptoms with high- and low-dose haloperidol: behavioral considerations. Arch Gen Psychiatry 1981; 38:195–199Crossref, Medline, Google Scholar
12. Lerner Y, Lwow E, Levitin A, Belmaker RH: Acute high-dose parenteral haloperidol treatment of psychosis. Am J Psychiatry 1979; 136:1061–1064Link, Google Scholar
13. Anderson WH, Kuehnle JC, Catanzano DM: Rapid treatment of acute psychosis. Am J Psychiatry 1976; 133:1076–1078Link, Google Scholar
14. Stern RG, Kahn RS, Harvey PD, Amin F, Apter SH, Hirschowitz J: Early response to haloperidol treatment in chronic schizophrenia. Schizophr Res 1993; 10:165–171Crossref, Medline, Google Scholar
15. Man PL, Chen CH: Rapid tranquilization of acutely psychotic patients with intramuscular haloperidol and chlorpromazine. Psychosomatics 1973; 14:59–63Crossref, Medline, Google Scholar
16. Glovinsky D, Kirch DG, Wyatt RJ: Early antipsychotic response to resumption of neuroleptics in drug-free chronic schizophrenic patients. Biol Psychiatry 1992; 31:968–970Crossref, Medline, Google Scholar
17. Wright P, Birkett M, David SR, Meehan K, Ferchland I, Alaka KJ, Saunders JC, Krueger J, Bradley P, San L, Bernardo M, Reinstein M, Breier A: Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry 2001; 158:1149–1151Link, Google Scholar
18. Kay SR, Sevy S: Pyramidical model of schizophrenia. Schizophr Bull 1990; 16:537–545Crossref, Medline, Google Scholar
19. Czobor P, Volavka J: Dimensions of the Brief Psychiatric Rating Scale: an examination of stability during haloperidol treatment. Compr Psychiatry 1996; 37:205–215Crossref, Medline, Google Scholar
20. Kraemer HC, Wilson GT, Fairburn CG, Agras WS: Mediators and moderators of treatment effects in randomized clinical trials. Arch Gen Psychiatry 2002; 59:877–883Crossref, Medline, Google Scholar
21. Milton GV, Jann MW: Emergency treatment of psychotic symptoms: pharmacokinetic considerations for antipsychotic drugs. Clin Pharmacokinet 1995; 28:494–504Crossref, Medline, Google Scholar
22. Bergstrom RF, Mitchell MI, Cerimele BJ, Hatcher BL, Jewell H, Satterwhite JH, Harry J: Direct comparison of im and oral olanzapine pharmacokinetics in healthy male subjects (abstract). AAPS PharmSci 1999; 1:3206Google Scholar
23. Cressman WA, Bianchine JR, Slotnick VB, Johnson PC, Plostnieks J: Plasma level profile of haloperidol in man following intramuscular administration. Eur J Clin Pharmacol 1974; 7:99–103Crossref, Medline, Google Scholar
24. Forsman A, Ohman R: Studies on serum protein binding of haloperidol. Curr Ther Res Clin Exp 1977; 21:245–255Medline, Google Scholar
25. Settle EC Jr, Ayd FJ Jr: Haloperidol: a quarter century of experience. J Clin Psychiatry 1983; 44:440–448Medline, Google Scholar
26. Tauscher J, Jones C, Remington G, Zipursky RB, Kapur S: Significant dissociation of brain and plasma kinetics with antipsychotics. Mol Psychiatry 2002; 7:317–321Crossref, Medline, Google Scholar
27. Nordstrom AL, Farde L, Halldin C: Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology (Berl) 1992; 106:433–438Crossref, Medline, Google Scholar
28. Wright P, Meehan K, Birkett M, Lindborg SR, Taylor CC, Morris P, Breier A: A comparison of the efficacy and safety of olanzapine versus haloperidol during transition from intramuscular to oral therapy. Clin Ther 2003; 25:1420–1428Crossref, Medline, Google Scholar
29. Bunney BS, Grace AA: Acute and chronic haloperidol treatment: comparison of effects on nigral dopaminergic cell activity. Life Sci 1978; 23:1715–1727Crossref, Medline, Google Scholar
30. Grace AA, Bunney BS: Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. J Pharmacol Exp Ther 1986; 238:1092–1100Medline, Google Scholar
31. Grace AA, Bunney BS, Moore H, Todd CL: Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 1997; 20:31–37Crossref, Medline, Google Scholar
32. Hyman SE, Nestler EJ: Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry 1996; 153:151–162Link, Google Scholar
33. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M: Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA 2000; 97:8104–8109Crossref, Medline, Google Scholar
34. Miller R: The time course of neuroleptic therapy for psychosis: role of learning processes and implications for concepts of psychotic illness. Psychopharmacology (Berl) 1987; 92:405–415Crossref, Medline, Google Scholar
35. Miller R: Major psychosis and dopamine: controversial features and some suggestions. Psychol Med 1984; 14:779–789Crossref, Medline, Google Scholar
36. Kapur S: Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 2003; 160:13–23Link, Google Scholar
37. Berridge KC, Robinson TE: What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 1998; 28:309–369Crossref, Medline, Google Scholar
38. Schultz W: Getting formal with dopamine and reward. Neuron 2002; 36:241–263Crossref, Medline, Google Scholar

