
Psychopathology in Patients With Degenerative Cerebellar Diseases: A Comparison to Huntington’s Disease
Abstract
OBJECTIVE: This study estimated the psychiatric morbidity of patients with degenerative cerebellar diseases. METHOD: The study included a series of 31 patients with degenerative cerebellar diseases, compared with 21 patients with Huntington’s disease and 29 neurologically healthy comparison subjects. Comprehensive psychiatric evaluations, including the Structured Clinical Interview for DSM-IV and psychopathology rating scales, were administered. RESULTS: The overall rate of noncognitive psychiatric disorders was 77% in the patients with degenerative cerebellar diseases, nearly identical to that in the patients with Huntington’s disease (81%) and about double that seen in the neurologically healthy subjects (41%). There were high rates of all mood disorders in both the degenerative cerebellar diseases group (68%) and the Huntington’s disease group (43%); the rate in the degenerative cerebellar diseases group was significantly higher than that in the neurologically healthy subjects (31%). The frequency of personality change in the three groups was striking: change was present in 26% of the degenerative cerebellar diseases patients, 48% of the Huntington’s disease patients, and none of the neurologically healthy comparison subjects. A total of 19% of the degenerative cerebellar diseases subjects and 71% of the Huntington’s disease subjects met DSM-IV criteria for either cognitive disorder or dementia. CONCLUSIONS: The high rate of psychiatric and cognitive disorders in the patients with degenerative cerebellar diseases suggests that many, if not most, patients with degenerative cerebellar diseases may benefit from psychiatric interventions. These results also support previous findings that the cerebellum may have a role in modulating emotion and cognition.
Over the past decade, a growing body of work has provided compelling support for nonmotor functions of the cerebellum. Evidence for this derives in part from elegant neuroanatomic studies demonstrating multiple parallel loops between the cerebellum and diverse regions of the cerebral cortex, including nonmotor limbic and frontal areas (1, 2), similar to the loops linking the basal ganglia to different regions of the cerebral cortex (3). Other pathways of potential relevance include reciprocal connections between the cerebellum and brainstem catecholaminergic (locus ceruleus) (4) and serotoninergic (dorsal raphe) nuclei (5), the hypothalamus (6), the brainstem reticular formation (7), and the ventral tegmental dopaminergic neurons projecting to the forebrain (8). These connections are of functional significance; investigations of normal human and nonhuman primate cerebellar function suggest a modulating role for the cerebellum in a variety of cognitive and emotional domains (9, 10).
Consistent with the existence of nonmotor cerebellar functions, preliminary evidence points to cognitive and psychiatric disturbances in patients with diseases affecting the cerebellum. For instance, most—but not all—investigations have found that subjects with moderate degenerative cerebellar diseases develop mild subcortical dementia (11, 12). Numerous case reports have suggested the presence of psychiatric disease in patients with cerebellar damage, including mania (13, 14), “psychosis” (15), and schizophrenia (16). Skre (17), examining subjects with any type of hereditary ataxia and using largely undefined and now obsolete psychiatric diagnostic schemes, found psychiatric disorders in approximately 23% of subjects, 12% of their neurologically normal family members, and 3% of the neurologically normal individuals from families without neurological disease. Elevated rates of short-lived mild depression have been detected acutely after cerebellar or brainstem strokes (18). Schmahmann and Sherman (19) examined 20 consecutive patients, primarily with cerebellar stroke, and described a “cerebellar cognitive affective syndrome” that involved executive, visuospatial, and linguistic dysfunction, accompanied by personality change.
Most recently, we reviewed the neurological records of 133 patients with cerebellar degeneration who were referred for genetic testing (20). The neurologists’ notes indicated that 41% of these patients had noncognitive psychopathology, particularly depression and personality change, and 30% were cognitively impaired. The patients with more basal ganglia involvement had higher rates of cognitive deficits and noncognitive psychiatric disorders.
The present study was designed to address the issue of cognitive and noncognitive psychopathology in degenerative cerebellar diseases, using standardized methods of assessment and both neurologically impaired and neurologically healthy comparison groups. In an attempt to minimize diagnostic heterogeneity, we chose patients with degenerative cerebellar disease, either spinocerebellar ataxia or multisystem atrophy, adult-onset progressive disorders that affect the cerebellum (21). Spinocerebellar ataxia is actually a large group of very similar hereditary and sporadic disorders (including so-called “pure cerebellar degeneration”) that are characterized by signs and symptoms of cerebellar dysfunction, frequently accompanied by evidence of involvement of other regions of the nervous system, and that are not secondary to other medical conditions such as stroke or cancer. The diagnosis of multisystem atrophy, usually considered a sporadic disorder, depends on evidence of combined involvement of the cerebellum, the basal ganglia, the pyramidal tracts, and the autonomic system—also unexplained by a medical condition. In practice, the phenotypes of spinocerebellar ataxia and multisystem atrophy overlap, and it is possible to select subjects with either diagnosis who have findings largely limited to the cerebellum.
We hypothesized that subjects with degenerative cerebellar diseases would have more frequent and more severe psychopathology than neurologically healthy comparison subjects. We also predicted that psychopathology would be less prominent in patients with degenerative cerebellar diseases than in a matched group of subjects with Huntington’s disease, a disorder characterized by basal ganglia degeneration, relative sparing of the cerebellum, and common and often severe psychopathology (22, 23). The results confirmed our first hypothesis that psychopathology is frequent in patients with degenerative cerebellar diseases. Contrary to expectations, psychopathology in patients with degenerative cerebellar diseases is nearly as common as in Huntington’s disease, although less severe and somewhat different in type.
Method
Subjects
Eighty-one adult subjects (age 18 and over) were ascertained through consecutive referrals from local neurologists, the Baltimore Huntington’s Disease Center, and members of the Chesapeake Chapter of the National Ataxia Foundation. The study group consisted of three subgroups of subjects: 31 with degenerative cerebellar diseases (Table 1), 21 with early- to mid-stage Huntington’s disease (confirmed by genetic testing and longitudinal clinical examinations), and 29 neurologically normal comparison subjects. Subjects in the degenerative cerebellar diseases group had a diagnosis of either spinocerebellar ataxia (including pure cerebellar degeneration) or multisystem atrophy with predominant cerebellar findings upon neurological examinations. A total of 17 of the 31 patients with degenerative cerebellar diseases had also been included in our previous chart review study. The diagnoses of degenerative cerebellar diseases or Huntington’s disease were confirmed by a neurologist specializing in movement disorders (E.O.). Only affected subjects with a minimum of 5 years of disease duration were included. Neurologically healthy comparison subjects were recruited from among spouses, other neurologically healthy relatives not at genetic risk for degenerative cerebellar diseases or Huntington’s disease, and caregivers of the patients with degenerative cerebellar diseases. Any subject with a current DSM-IV diagnosis of substance use or dependence or using drugs or alcohol in the 48 hours before examination was excluded. On the basis of these criteria, we excluded one patient with degenerative cerebellar diseases and one neurologically healthy subject. Other exclusion criteria included metabolic or toxic factors known to affect the cerebellum, such as long-term phenytoin use, vitamin B12 deficiency, or untreated thyroid disease. No subjects were excluded for these reasons. One patient with degenerative cerebellar diseases was too cognitively impaired to complete the test battery and was excluded. After a complete description of the study was given to the subjects, written informed consent was obtained in accordance with the guidelines of the Johns Hopkins Internal Review Board. Sociodemographic and clinical data were obtained from interviews and medical records.
Neurological Evaluation
The Quantitated Neurologic Examination (24), a well-established scale for rating the severity of Huntington’s disease, and the International Cooperative Ataxia Rating Scale (25) were used to provide quantitative measures of neurological symptom severity. The scores on the Quantitated Neurologic Examination and the International Cooperative Ataxia Rating Scale were combined and redundant items eliminated to render a total modified neurological score as a measure of overall neurological symptom severity.
Psychiatric Evaluation
Each subject in this study underwent a comprehensive psychiatric assessment by a neuropsychiatrist experienced in the assessment and treatment of patients with movement disorders (I.L.). The interview included present and past complaints; personal, social, and family history; and a mental state examination. In addition, the nonpatient version of the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID) (26) was administered to each subject. The SCID assumes that subjects are not identified as psychiatric patients, and no assumption about a chief complaint is made. An informant for each subject supplied further collateral information that was used to make a clinical diagnosis. Informants were specifically asked about distinct and enduring changes in behavior and personality since the onset of movement disorder or, for comparison subjects, over recent years. The data were reviewed by a second neuropsychiatrist (R.L.M.), who was blind to the neurological diagnosis, and a consensus diagnosis was established. For subjects with more than one axis I diagnosis, the principal diagnosis was the condition that was the main focus of attention or treatment at the time of evaluation. In reviewing past and present psychiatric history in the degenerative cerebellar diseases and Huntington’s disease groups, we made a distinction between psychiatric complaints before and after the onset of neurological symptoms. For the neurologically healthy comparison group, a distinction was made between lifetime psychiatric history and present psychiatric complaints.
For the purposes of analysis, brief recurrent depressive disorder, minor depressive disorder, and dysthymia were grouped together as “nonmajor depression.” Subjects with both major and nonmajor depression were coded only as having “major depression” to avoid overestimating the overall rate of depression in our group. A diagnosis of “personality change” was made when a persistent personality disturbance was clinically evident or reported by informants. All DSM-IV subtypes of personality disorder were considered: labile, disinhibited, aggressive, apathetic, and paranoid. In addition, we sought evidence for persistent personality disturbance not falling into these DSM-IV categories. In all patients diagnosed with personality change, the changes were severe enough to cause clinically important distress or impairment in social, occupational, or other important areas of functioning. To avoid the assumption of a causal relationship between personality change and cognitive impairment or dementia, we made the diagnosis of personality change independently of the diagnosis of dementia or cognitive impairment. Subjects could therefore have either a personality change, cognitive impairment or dementia, or both. The diagnosis of dementia was made in subjects with a Mini-Mental State Examination (MMSE) (27) score <24, evidence of cognitive impairment on informant history, and functional impairment. Cognitive impairment not otherwise specified was applied to subjects meeting the latter two criteria but with an MMSE ≥24. Cognitive and functional impairment was assessed by specific inquiries of both the subject and the informant regarding deterioration in job performance, activities of daily living (such as bathing, dressing, and toileting), and instrumental activities of daily living (such as shopping, telephone use, and managing finances).
Current level of psychopathology was obtained with the administration of specific clinician-rated scales: Hamilton Depression Rating Scale (28), Hamilton Anxiety Rating Scale (29), Young Mania Rating Scale (30), and the Yale-Brown Obsessive Compulsive Scale (31). An informant assessment scale to evaluate the presence and severity of apathy and irritability (32) was used. Each subject was also administered the National Adult Reading Test (33), to serve as a rough index of premorbid intellectual capacity, and a battery of neuropsychological tests described previously (unpublished data of Brandt et al.).
Analysis
Statistical analyses were performed with the Statistical Package for the Social Sciences (34). We used chi-square tests, t tests, or one-way analyses of variance, with Scheffé post hoc analyses for comparison of each pair of groups. Because of the relatively small group size and to avoid a type II error while minimizing type I error, we chose a conservative alpha level (p<0.02) rather than an a priori correction for multiple comparisons, which might have failed to identify a real effect.
Results
Demographic and Neurological Characteristics
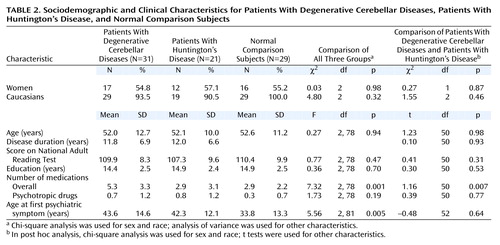
Age, sex, race, National Adult Reading Test scores, and education level were similar among the three groups (Table 2). Duration of disease was similar in the Huntington’s disease and degenerative cerebellar diseases groups. Number of medicines was significantly higher in the degenerative cerebellar diseases group than in the other groups, but there was no difference in the number of psychotropic medicines among the groups. The mean highest education level (about 15 years) as well as the predominantly Caucasian composition of the group suggests an ascertainment bias consistent across all three groups. The neurological patient groups were comparably matched on neurological deficits (Table 3).
Psychiatric Features
Psychiatric diagnoses
On the basis of both the SCID scores and the psychiatric interviews and with use of DSM-IV criteria, significant differences among the three groups in the post-disease-onset or lifetime rate of mood disorders, anxiety disorders (excluding phobia), and psychotic disorders were observed (Figure 1). When we considered all axis I diagnoses other than cognitive impairment and dementia, the overall rate of psychiatric disorders was 77% in the degenerative cerebellar diseases group, which was nearly identical to that in the Huntington’s disease group (81%) (χ2=0.09, df=1, p=0.76) and nearly twice the rate in the neurologically healthy comparison group (41%) (χ2=8.11, df=1, p=0.004). In addition, in both patient groups, the mean age at onset of psychiatric symptoms was significantly later than the mean age at onset of psychiatric symptoms in the neurologically normal group (Table 2). Significant psychopathology was present before the onset of motor symptoms in only one subject with degenerative cerebellar diseases. No significant gender differences in the frequency of psychiatric diagnoses were detected in any of the three groups.
The most common DSM diagnosis in the patient groups was personality change (25.8% in the degenerative cerebellar diseases group and 46.7% in the Huntington’s disease group) (χ2=2.63, df=1, p=0.11). Personality change was significantly more frequent in the two patient groups than in the neurologically healthy subjects (χ2=16.35, df=2, p<0.001), in whom personality change was not detected. The type of personality change in the patients with degenerative cerebellar diseases and Huntington’s disease was variable and included the DSM-IV subtypes labile, disinhibited, apathetic, and paranoid as well as a subtype, not described in DSM-IV, characterized by child-like, regressive behavior. Of note is that personality changes could not be completely explained by a generalized dementing process. A total of 50% of the subjects with degenerative cerebellar diseases and 20% of the Huntington’s disease subjects with personality change did not meet DSM-IV criteria for cognitive disorder or dementia.
A higher percentage of the neurological patients than the normal comparison subjects had a history of any mood disorder (57.7% versus 31%) (χ2=5.30, df=1, p=0.021). The frequency of a history of mood disorder was high in both degenerative cerebellar diseases and Huntington’s disease groups; the difference between the two groups did not reach statistical significance (67.7% versus 42.8%) (χ2=3.18, df=1, p=0.07). The rate of mood disorders in the degenerative cerebellar diseases group was significantly higher than in the neurologically normal comparison group (31%) (χ2=8.41, df=2, p<0.02). The same relative frequencies of mood disorder among the three groups were apparent when “mood disorder” was split into major depression (35.5% for the degenerative cerebellar diseases group, 28.6% for the Huntington’s disease group, and 17.2% for the normal comparison group) and nonmajor depression (32.3%, 14.3%, and 13.8%, respectively). Bipolar disorder or mania was not seen in any patient with degenerative cerebellar diseases but was diagnosed in one Huntington’s disease subject (4.8%). Within the degenerative cerebellar diseases group, there was no association between mood disorder and total neurological severity score, ataxia score, duration of illness, education level, or MMSE score.
Current psychopathology
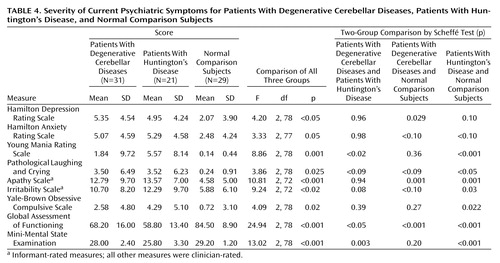
Each subject was rated with a variety of instruments designed to establish the severity of current psychopathology (Table 4). Overall scores on these rating instruments tended to be fairly low, with the exception of apathy and irritability. This is consistent with our clinical observation that only a few subjects were acutely ill at the time of the examination. Nonetheless, a difference among the three groups approaching or reaching the <0.02 level of significance was apparent on all measures, with both neurological patient groups scoring higher than the neurologically healthy comparison group and similar to each other. These findings confirm the high rate of personality change in the degenerative cerebellar diseases and Huntington’s disease groups, with prominent features of apathy, irritability, and emotional lability. There was also preliminary evidence evident in most of the ratings and in the Global Assessment of Functioning Scale that, on average, psychopathology may be more severe in patients with Huntington’s disease than in patients with degenerative cerebellar diseases.
Clinically detectable cognitive disorders
The three groups were well matched on premorbid IQ on the basis of the National Adult Reading Test (Table 2). Among the neurologic patients, 19.4% of the subjects with degenerative cerebellar diseases and 71.4% of the Huntington’s disease subjects (χ2=14.00, df=1, p<0.001) were either demented or more mildly cognitively impaired on the basis of modified DSM-IV criteria (three patients with degenerative cerebellar diseases were assigned to each category; eight Huntington’s disease patients were demented, and seven were cognitively impaired). This finding, in conjunction with a nearly identical duration of illness across groups and similar levels of neurological symptoms, suggests that degenerative cerebellar diseases are less cognitively impairing than Huntington’s disease. The mean MMSE score of the Huntington’s disease group was significantly lower than the mean MMSE of both the degenerative cerebellar diseases patients and the neurologically healthy comparison group (Table 4).
Discussion
The primary result of this study was that psychiatric manifestations of degenerative cerebellar diseases were detectable in 77% of the subjects, similar to the rate observed in Huntington’s disease and significantly higher than the rate of psychiatric disorders seen in neurologically normal comparison subjects. The most common diagnoses in the patients with degenerative cerebellar diseases were depressive disorders, personality changes, and cognitive impairment. The rate of noncognitive psychopathology was higher than in our chart review study, while the rate of clinically important cognitive impairment was lower. The former difference is an expected consequence of the nature of the assessments in each study. The latter was somewhat surprising and may indicate that the criteria employed in the present analysis were more conservative than those applied during routine neurological examinations or showed a difference in subject ascertainment between the two studies. The comparable overall rates of noncognitive psychopathology in the patients with degenerative cerebellar diseases and the patients with Huntington’s disease were also somewhat surprising. However, the overall rates did not provide a complete picture, since the Huntington’s disease patients tended to have more severe manifestations of the disorders and the rate of cognitive impairment was substantially higher in the patients with Huntington’s disease.
Although a history of mood disorders was common in the patients with degenerative cerebellar diseases, our cross-sectional evaluation indicated that few patients with degenerative cerebellar diseases were depressed at the time of the evaluation, similar to the findings of Kish et al. (12). The low rate of cross-sectional depression confirms the importance of a longitudinal psychiatric history in assessing psychopathology in patients with degenerative cerebellar diseases and implies that degenerative cerebellar diseases predispose to intermittent rather than chronic mood disorders. Bipolar disorder or mania was not seen in our degenerative cerebellar diseases group, in contrast to the “incidental observations” of Kish et al. (12) that 14% of the patients with degenerative cerebellar diseases were “mildly euphoric” and 12% were “impulsive,” and the report of Lauterbach (13) that 20% of subjects with focal cerebellar circuit lesions developed bipolar disorders. These differences likely derive from variability in patient populations and diagnostic protocols and the relatively small size of all samples.
Personality change was a frequent diagnosis in both patients with Huntington’s disease and patients with degenerative cerebellar diseases. These changes were difficult to detect with rating scales but did emerge on careful clinical evaluation. A total of 50% of the patients with degenerative cerebellar diseases and 20% of the patients with Huntington’s disease with personality change did not meet the criteria for cognitive impairment or dementia, suggesting that personality changes may be an independent manifestation of neurodegeneration rather than a nonspecific aspect of a dementing process. The personality changes in our subjects were similar to the affective component of the cognitive affective syndrome described by Schmahmann and Sherman (19). However, the design of our study enabled us to demonstrate that degenerative cerebellar diseases predispose patients to a variety of psychiatric disorders rather than a single affective syndrome. The extent to which these disorders are also present in patients with focal cerebellar lesions, such as strokes, remains to be determined.
Our method for determining personality change was based on a DSM-IV-based interview, an informant history of personality change, and cross-sectional informant rating scales of irritability and apathy. Other investigators have applied standardized quantitative rating scales of personality dimensions, such as the Neuroticism-Extroversion-Openness Personality Inventory, to Alzheimer’s disease and other neuropsychiatric disorders (35–37), with informants providing separate ratings of premorbid and current personality. The reliability of the Neuroticism-Extroversion-Openness Personality Inventory to detect personality change in Alzheimer’s disease has been preliminarily established (38, 39). However, the extent to which the personality dimensions assessed in the Neuroticism-Extroversion-Openness Personality Inventory correlate with syndromes typically associated with neuropsychiatric disorders (e.g., apathy, irritability, and agitation) remains to be fully established. Other standardized assessments have attempted to directly approach personality variables in neuropsychiatric patients from a categorical (syndromic) perspective (40–42); the reliability of these instruments in detecting personality change, particularly in patients with degenerative cerebellar diseases, is not known. It is possible that use of one or more of these more fully operationalized diagnostic procedures in our study, whether dimensional or categorical, may have yielded different findings concerning personality change in patients with degenerative cerebellar diseases and Huntington’s disease. More generally, however, the analysis and validation of personality change in neuropsychiatric conditions are clearly in a nascent stage. The vagueness of the DSM-IV personality change diagnosis (“personality change due to a general medical condition,” 310.1), the unclear relationship between personality change and cognitive impairment, and, most significant, conceptual uncertainty regarding classification and description of personality change (36) create limitations in interpreting the current data.
One case of schizophrenia and two cases of psychotic disorder not otherwise specified were detected in the group with degenerative cerebellar diseases but none in either of the two other groups. The presence of these syndromes in 10% of our subjects is high enough to suggest that cerebellar degeneration may increase the risk for psychotic phenomena, consistent with the concept of “cognitive dysmetria” proposed by Schmahmann (43) and applied to schizophrenia by Andreasen and colleagues (44). The essence of this hypothesis is that the fluidity and synchrony of thought, like movement, are normally modulated by the cerebellum. Hence, if circuits between the cerebellum and cerebral cortex develop abnormally or suffer damage, the result may be misperception and cognitive dysfunction. In support of this hypothesis, anatomical and functional neuroimaging studies have suggested cerebellar involvement in idiopathic schizophrenia and affective disorder, although the evidence remains controversial (44–46).
Almost 20% of the patients with degenerative cerebellar diseases and over 70% of the patients with Huntington’s disease had functionally significant cognitive disorders. It is unlikely that the cognitive impairment that we observed can be completely explained by high rates of depression, since cross-sectional ratings of depression in our subjects were relatively low at the time of the assessment. These clinical results are consistent with our preliminary analysis of a detailed neuropsychological evaluation of the same subjects (unpublished data of Brandt et al.). Subjects with degenerative cerebellar diseases appear most impaired in executive function and least impaired in memory, whereas the greatest impairments in the subjects with Huntington’s disease were in measures of spatial ability and memory. Our preliminary chart review suggestion of a 30% rate of cognitive impairment in subjects with degenerative cerebellar diseases was higher than the results of the current study or of previous reports, reflecting a difference in the study methods or subject population.
The extent to which the psychopathology of degenerative cerebellar diseases reflects a primary manifestation of neuropathology, as opposed to a secondary and nonspecific manifestation of the demoralization of chronic illness, is a critical issue. The lower rates of psychopathology in the group of neurologically normal comparison subjects, chosen because of their exposure to the psychological stress of chronic illness in their role as caregivers and family members, provide evidence that the psychopathology of degenerative cerebellar diseases is directly linked to brain pathology. This link is also supported by the syndromic nature of the psychiatric disorders detected in patients with degenerative cerebellar diseases and particularly the presence of personality changes and psychotic disorders. In addition, although Huntington’s disease and degenerative cerebellar diseases groups both exhibited the features typical of subcortical dementia, the cognitive and psychopathological manifestations of the two diseases were not identical. Since the illness duration and motor impairment in each group were similar, this phenomenological difference appears to reflect the underlying neuropathology of each disease. Recent work has established that the regions of the cerebral cortex in direct communication with the cerebellum only partially overlap with these regions communicating with the basal ganglia. For instance, projections to prefrontal area 12, roughly corresponding to the orbital cortex, have been detected from the basal ganglia but not from the cerebellothalamic system (3). Alternatively, differences in psychopathology may be a consequence of the different modes of information processing in the basal ganglia and cerebellum, an attractive hypothesis given the unique modular nature of cerebellar microcircuitry (47).
Aside from concerns regarding the diagnosis and interpretation of personality change, other important limitations of this study include the modest group size, the cross-sectional design, the inherent subjectivity of psychiatric diagnoses, and the heterogeneity of the group with degenerative cerebellar diseases. The etiology of the degenerative cerebellar diseases was variable, and in many subjects, neuropathology may have extended beyond the confines of the cerebellum, although predominant cerebellar findings were the basis for subject selection. While noncerebellar neuropathology increases the difficulty of interpreting the specific role of the cerebellum in our findings, a strength of the study group was that it more closely resembled the population of patients with degenerative cerebellar disease that are seen for clinical care than a group with disease limited to the cerebellum.
In summary, we reported high rates of psychiatric disorders and clinically detectable cognitive impairment in 31 subjects with degenerative cerebellar diseases. Within the limitations of the study design, the results support a role for the cerebellum in the modulation of cognition and emotion. From a clinical perspective, our findings suggest that patients with degenerative cerebellar diseases should be carefully evaluated for the presence of psychiatric disorders and cognitive impairments. Although the underlying neurodegeneration in degenerative cerebellar diseases may not yet be treatable, management of the accompanying psychiatric disorders and cognitive impairment with a combination of education, pharmacotherapy, and supportive psychotherapy may have a major impact on quality of life for patients and their families.
 |
 |
 |
 |
Presented in part at the 12th annual meeting of the American Neuropsychiatric Association, Fort Myers, Fla., Feb. 24–27, 2001; and at the Mental and Behavioral Dysfunction in Movement Disorders International Symposium, Montreal, Oct. 10–13, 2001. Received Sept. 6, 2001; revisions received Dec. 28, 2001, and Feb. 21, 2002; accepted March 5, 2002. From the Program in Cellular and Molecular Medicine and the Departments of Psychiatry and Behavioral Sciences, Neurology, and Neuroscience, Johns Hopkins University School of Medicine. Address reprint requests to Dr. Margolis, Johns Hopkins University School of Medicine, Meyer 2-181, 600 N. Wolfe St., Baltimore, MD 21287; [email protected] (e-mail). Supported by an Independent Investigator Award from the National Association for Research in Schizophrenia and Affective Disorders (Dr. Margolis) and NIH grants RR-00052 (from the Division of Research Resources), NS-16375, and NS-35872 (from the National Institute of Neurological and Communicative Disorders and Stroke). The authors thank Drs. Godfrey Pearlson, Elizabeth Aylward, Stephen Reich, Stephen Grill, H.A. Jinnah, and Paul R. McHugh, the members of the Chesapeake Chapter of the National Ataxia Foundation, Christine Liszewski, Debbie Pollard, Marie Sonderman, Alvaro Bilbao, Kate Tracy, and Barnett Shpritz for their help.

Figure 1. Frequency of Psychiatric Disordersa in Patients With Degenerative Cerebellar Diseases, Patients With Huntington’s Disease, and Normal Comparison Subjects
aMood disorders included major depression, brief recurrent depression, minor depressive disorder, dysthymia, and bipolar disorder—mania. Diagnoses of personality change were made independent of cognitive status. Anxiety disorders included generalized anxiety disorder, panic disorder, obsessive-compulsive disorder, phobia, and posttraumatic stress disorder. Psychotic disorders included schizophrenia and psychotic disorder not otherwise specified. Chi-square tests were used for all analyses.
bSignificant difference from normal comparison subjects (χ2=8.01, df=1, p=0.004).
cSignificant difference from normal comparison subjects (χ2=8.64, df=1, p=0.003).
dSignificant difference from normal comparison subjects (χ2=17.26, df=1, p<0.001).
eSignificant difference from normal comparison subjects (χ2=8.11, df=1, p=0.004).
fSignificant difference from normal comparison subjects (χ2=7.83, df=1, p=0.005).
gSignificant difference across all three groups (χ2=8.41, df=2, p<0.02).
hSignificant difference across all three groups (χ2=16.35, df=2, p<0.001).
iSignificant difference across all three groups (χ2=11.62, df=2, p=0.003).
1. Schmahmann JD, Pandya DN: The cerebrocerebellar system. Int Rev Neurobiol 1997; 41:31-60Crossref, Medline, Google Scholar
2. Middleton FA, Strick PL: Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev 2000; 31:236-250Crossref, Medline, Google Scholar
3. Alexander GE, Crutcher MD, DeLong MR: Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res 1990; 85:119-146Crossref, Medline, Google Scholar
4. Dempesy CW, Tootle DM, Fontana CJ, Fitzjarrell AT, Garey RE, Heath RG: Stimulation of the paleocerebellar cortex of the cat: increased rate of synthesis and release of catecholamines at limbic sites. Biol Psychiatry 1983; 18:127-132Medline, Google Scholar
5. Marcinkiewicz M, Morcos R, Chretien M: CNS connections with the median raphe nucleus: retrograde tracing with WGA-apoHRP-Gold complex in the rat. J Comp Neurol 1989; 289:11-35Crossref, Medline, Google Scholar
6. Haines DE, Dietrichs E, Mihailoff GA, McDonald EF: The cerebellar-hypothalamic axis: basic circuits and clinical observations. Int Rev Neurobiol 1997; 41:83-107Crossref, Medline, Google Scholar
7. Brodal A: Neurological Anatomy. New York, Oxford University Press, 1981Google Scholar
8. Snider RS, Maiti A, Snider SR: Cerebellar pathways to ventral midbrain and nigra. Exp Neurol 1976; 53:714-728Crossref, Medline, Google Scholar
9. Berman AJ: Amelioration of aggression: response to selective cerebellar lesions in the rhesus monkey. Int Rev Neurobiol 1997; 41:111-119Crossref, Medline, Google Scholar
10. Liotti M, Mayberg HS, Brannan SK, McGinnis S, Jerabek P, Fox PT: Differential limbic-cortical correlates of sadness and anxiety in healthy subjects: implications for affective disorders. Biol Psychiatry 2000; 48:30-42Crossref, Medline, Google Scholar
11. Kish SJ, El-Awar M, Stuss D, Nobrega J, Currier R, Aita JF, Schut L, Zoghbi HY, Freedman M: Neuropsychological test performance in patients with dominantly inherited spinocerebellar ataxia: relationship to ataxia severity. Neurology 1994; 44:1738-1746Crossref, Medline, Google Scholar
12. Kish SJ, el Awar M, Schut L, Leach L, Oscar-Berman M, Freedman M: Cognitive deficits in olivopontocerebellar atrophy: implications for the cholinergic hypothesis of Alzheimer’s dementia. Ann Neurol 1988; 24:200-206Crossref, Medline, Google Scholar
13. Lauterbach EC: Bipolar disorders, dystonia, and compulsion after dysfunction of the cerebellum, dentatorubrothalamic tract, and substantia nigra. Biol Psychiatry 1996; 40:726-730Crossref, Medline, Google Scholar
14. Cutting JC: Chronic mania in childhood: case report of a possible association with a radiological picture of cerebellar disease. Psychol Med 1976; 6:635-642Crossref, Medline, Google Scholar
15. Kutty IN, Prendes JL: Psychosis and cerebellar degeneration. J Nerv Ment Dis 1981; 169:390-391Crossref, Medline, Google Scholar
16. Tashiro H, Suzuki SO, Hitotsumatsu T, Iwaki T: An autopsy case of spinocerebellar ataxia type 6 with mental symptoms of schizophrenia and dementia. Clin Neuropathol 1999; 18:198-204Medline, Google Scholar
17. Skre H: A study of certain traits accompanying some inherited neurological disorders. Clin Genet 1975; 8:117-135Crossref, Medline, Google Scholar
18. Starkstein SE, Robinson RG, Berthier ML, Price TR: Depressive disorders following posterior circulation as compared with middle cerebral artery infarcts. Brain 1988; 111:375-387Crossref, Medline, Google Scholar
19. Schmahmann JD, Sherman JC: Cerebellar cognitive affective syndrome. Int Rev Neurobiol 1997; 41:433-440Crossref, Medline, Google Scholar
20. Leroi I, O’Hearn E, Margolis RL: Psychiatric syndromes in cerebellar degeneration. Int Rev Psychiatry 2001; 13:323-329Crossref, Google Scholar
21. Shill HA, Hallet M: Cerebellar diseases. Int Rev Psychiatry 2001; 13:261-267Crossref, Google Scholar
22. Folstein SE: Huntington’s Disease: A Disorder of Families. Baltimore, Johns Hopkins University Press, 1989Google Scholar
23. Ross CA, Margolis RL, Rosenblatt A, Ranen NG, Becher MW, Aylward E: Reviews in molecular medicine: Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine 1997; 76:305-338Crossref, Medline, Google Scholar
24. Folstein SE, Jensen B, Leigh RJ, Folstein MF: The measurement of abnormal movement: methods developed for Huntington disease. Neurobehav Tox Teratol 1983; 5:605-609Medline, Google Scholar
25. Trouillas P, Takayanagi T, Hallett M, Currier RD, Subramony SH, Wessel K, Bryer A, Diener HC, Massaquoi S, Gomez CM, Coutinho P, Ben Hamida M, Campanella G, Filla A, Schut L, Timann D, Honnorat J, Nighoghossian N, Manyam B (Ataxia Neuropharmacology Committee of the World Federation of Neurology): International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. J Neurol Sci 1997; 145:205-211Crossref, Medline, Google Scholar
26. First MB, Spitzer RL, Gibbon M, Williams JB: Structured Clinical Interview for DSM-IV Axis I Disorders—Non-Patient Edition (SCID-I/NP), version 2.0. New York, New York State Psychiatric Institute, Biometrics Research, 1996Google Scholar
27. Folstein MF, Folstein SE, McHugh PR: “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189-198Crossref, Medline, Google Scholar
28. Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56-62Crossref, Medline, Google Scholar
29. Hamilton M: The assessment of anxiety states by rating. Br J Med Psychol 1959; 32:50-55Crossref, Medline, Google Scholar
30. Young RC, Biggs JT, Ziegler VE, Meyer DA: A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry 1978; 133:429-435Crossref, Medline, Google Scholar
31. Goodman WK, Price LH, Rasmussen SA, Mazure C, Fleischmann RL, Hill CL, Heninger GR, Charney DS: The Yale-Brown Obsessive Compulsive Scale, I: development, use, and reliability. Arch Gen Psychiatry 1989; 46:1006-1011Crossref, Medline, Google Scholar
32. Burns A, Folstein S, Brandt J, Folstein M: Clinical assessment of irritability, aggression, and apathy in Huntington and Alzheimer disease. J Nerv Ment Dis 1990; 178:20-26Crossref, Medline, Google Scholar
33. Blair JR, Spreen O: Predicting premorbid IQ: a revision of the National Adult Reading Test. Clin Neuropsychol 1989; 3:129-136Crossref, Google Scholar
34. Statistical Products and Service Solutions 2000: Graduate Pack Advanced Version 10. Chicago, SPSS, 2000Google Scholar
35. Siegler IC, Welsh KA, Dawson DV, Fillenbaum GG, Earl NL, Kaplan EB, Clark CM: Ratings of personality change in patients being evaluated for memory disorders. Alzheimer Dis Assoc Disord 1991; 5:240-250Crossref, Medline, Google Scholar
36. Chatterjee A, Strauss ME, Smyth KA, Whitehouse PJ: Personality change in Alzheimer’s disease. Arch Neurol 1992; 49:486-491Crossref, Medline, Google Scholar
37. Dawson DV, Welsh-Bohmer KA, Siegler IC: Premorbid personality predicts level of rated personality change in patients with Alzheimer disease. Alzheimer Dis Assoc Disord 2000; 14:11-19Crossref, Medline, Google Scholar
38. Strauss ME, Pasupathi M, Chatterjee A: Concordance between observers in descriptions of personality change in Alzheimer’s disease. Psychol Aging 1993; 8:475-480Crossref, Medline, Google Scholar
39. Strauss ME, Pasupathi M: Primary caregivers’ descriptions of Alzheimer patients’ personality traits: temporal stability and sensitivity to change. Alzheimer Dis Assoc Disord 1994; 8:166-176Crossref, Medline, Google Scholar
40. Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J: The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994; 44:2308-2314Crossref, Medline, Google Scholar
41. Grace J, Stout JC, Malloy PF: Assessing frontal lobe behavioral syndromes with the Frontal Lobe Personality Scale. Assessment 1999; 6:269-284Crossref, Medline, Google Scholar
42. Kertesz A, Nadkarni N, Davidson W, Thomas AW: The Frontal Behavioral Inventory in the differential diagnosis of frontotemporal dementia. J Int Neuropsychol Soc 2000; 6:460-468Crossref, Medline, Google Scholar
43. Schmahmann JD: An emerging concept: the cerebellar contribution to higher function. Arch Neurol 1991; 48:1178-1187Crossref, Medline, Google Scholar
44. Andreasen NC, Nopoulos P, O’Leary DS, Miller DD, Wassink T, Flaum M: Defining the phenotype of schizophrenia: cognitive dysmetria and its neural mechanisms. Biol Psychiatry 1999; 46:908-920Crossref, Medline, Google Scholar
45. Rapoport M, van Reekum R, Mayberg H: The role of the cerebellum in cognition and behavior: a selective review. J Neuropsychiatry Clin Neurosci 2000; 12:193-198Crossref, Medline, Google Scholar
46. Katsetos CD, Hyde TM, Herman MM: Neuropathology of the cerebellum in schizophrenia—an update: 1996 and future directions. Biol Psychiatry 1997; 42:213-224Crossref, Medline, Google Scholar
47. O’Hearn E, Molliver M: Organizational principles and microcircuitry of the cerebellum. Int Rev Psychiatry 2001; 13:232-246Crossref, Google Scholar

