
Inhibition of Norepinephrine Uptake in Patients With Major Depression Treated With Paroxetine
Abstract
OBJECTIVE: The study examined whether paroxetine inhibits the human norepinephrine transporter in addition to the human serotonin (5-HT) transporter in patients with major depressive disorder. METHOD: In an open-label, parallel-group, forced-titration study, 52 outpatients with DSM-IV major depressive disorder and a baseline Montgomery Åsberg Depression Rating Scale score ≥20 were randomly assigned to treatment with paroxetine (to 60 mg/day) or desipramine (to 30 mg/day) in a 3-to-1 ratio, respectively. Norepinephrine and 5-HT transporter function were assayed by using human transporter transfected cells in the presence of serum collected at baseline and the end of each treatment week. Data from 36 patients were analyzed. RESULTS: Paroxetine decreased norepinephrine uptake to 73% of control (27% inhibition) at an average serum concentration of 100 ng/ml and 57% of control (43% inhibition) at 200 ng/ml. Uptake of 5-HT was decreased to less than 15% (greater than 85% inhibition) of control at these paroxetine concentrations. Desipramine decreased norepinephrine uptake to near maximal 15% of control (85% inhibition) at 100 ng/ml. Uptake of 5-HT was decreased to 82% of control (18% inhibition) at 100 ng/ml and 49% of control (51% inhibition) at 500 ng/ml. CONCLUSIONS: Paroxetine, currently classified as a selective 5-HT reuptake inhibitor, can act as a 5-HT/norepinephrine uptake inhibitor in vivo. The clinical significance of this action on norepinephrine uptake is currently unknown, but this action may contribute to the broad therapeutic efficacy of paroxetine in the treatment of depression, panic disorder, social anxiety disorder, posttraumatic stress disorder, and generalized anxiety disorder.
Many of the antidepressants used to treat major depressive disorder are antagonists at the human norepinephrine transporter and/or human serotonin (5-HT) transporter (1–3). The percentage of patients responding to either selective noradrenergic reuptake inhibitors (including heterocyclic antidepressants such as maprotiline or desipramine) or 5-HT reuptake inhibitors (SSRIs) (including fluoxetine, citalopram, sertraline, paroxetine, or fluvoxamine) is approximately 60% (4). A lower percentage of patients treated with these agents return to their premorbid level of functioning, i.e., experience true remission.
Continued study of the norepinephrine and 5-HT neurotransmitter systems in relation to the actions of antidepressants has revealed that using either a combination of medications that inhibit the norepinephrine transporter and the 5-HT transporter or a single medication that inhibits both transporters may represent a more effective antidepressant treatment than a single selective treatment, particularly for severe or refractory depression (5, 6). In an open-label study, combination treatment with desipramine, which is a potent noradrenergic reuptake inhibitor, and the SSRI fluoxetine led to a more rapid onset of antidepressant efficacy than either treatment alone (7). Similarly, a combination of nortriptyline and fluoxetine or sertraline was more effective than the use of one of these agents alone in ameliorating treatment-resistant depression (8). However, combination treatment regimes are not problem-free, as the potential for drug-drug interactions may increase and the likelihood of treatment compliance decrease.
Venlafaxine is the first newer-generation antidepressant to be classified as a selective dual uptake or 5-HT/norepinephrine reuptake inhibitor because it inhibited the norepinephrine transporter in addition to the 5-HT transporter at higher doses in preclinical animal studies (9). The classic tricyclic antidepressants amitriptyline and imipramine are known to modulate both noradrenergic and serotonergic systems (10), but a drug with the 5-HT/norepinephrine reuptake inhibitor classification does not possess the anticholinergic, antihistaminergic, and antiadrenergic properties of tricyclic antidepressants. Evidence from a meta-analytic study indicated that at higher doses venlafaxine is more effective than more selective agents such as fluoxetine in bringing about remission in depressed patients (6).
Paroxetine has been classified as an SSRI on the basis of its very high affinity for the 5-HT transporter (Ki=0.065 nmol/liter), although evidence from several laboratories has indicated that paroxetine also shows moderate affinity for the norepinephrine transporter (Ki ∼40–85 nmol/liter) (11–14). Animal studies have confirmed that after systemic administration, paroxetine partly inhibits the norepinephrine transporter in rat brain homogenates (15). It is important to note that both the in vitro and the in vivo data demonstrate that paroxetine binds to and inhibits the norepinephrine transporter in a concentration-dependent manner. In the rat, serum paroxetine concentrations ranging from 100 to 500 ng/ml (mean=210 ng/ml) inhibited the norepinephrine transporter an average of 21%, while serum concentrations of more than 500 ng/ml resulted in an inhibition of 34%. Therefore, paroxetine, like venlafaxine, may act as a 5-HT/norepinephrine reuptake inhibitor at higher doses or serum concentrations.
On the basis of these preliminary data, we hypothesized that paroxetine would inhibit the norepinephrine transporter, in addition to the 5-HT transporter, at higher serum concentrations in patients with a diagnosis of major depression. This hypothesis was tested in a modified monoamine uptake assay by measuring uptake inhibition of norepinephrine and 5-HT in serum samples taken from patients treated with escalating doses of paroxetine or desipramine. This method maintains the important equilibrium between free and serum-protein-bound drug, hence modeling in vivo conditions where only free drug is accessible to the brain and clinically relevant sites of action. The results from these experiments clearly demonstrate that paroxetine inhibits the norepinephrine transporter at serum concentrations over 100 ng/ml, which are commonly achieved by means of clinically relevant dosing in patients with major depression.
Method
Study Design
This was a multicenter, open-label, parallel-group, forced-titration study of paroxetine or desipramine in patients with a diagnosis of major depressive disorder. Patients were recruited by physicians at six outpatient centers. Medication assignment was determined by means of a treatment assignment sheet provided to each center such that for every four patients assigned to study treatment, three were assigned to receive paroxetine and one was assigned to receive desipramine. The 7-week dosing schedule for paroxetine was 10 mg/day for 1 week, 20 mg/day for 2 weeks, 40 mg/day for 2 weeks, and 60 mg/day for 2 weeks. The 7-week dosing schedule for desipramine was 50 mg/day for 1 week, 100 mg/day for 2 weeks, 200 mg/day for 2 weeks, and 300 mg/day for 2 weeks.
Safety assessments were performed at each visit, and efficacy assessments were performed at weeks 2, 3, 5, and 7 or at dropout, if applicable. For patients assigned to receive desipramine, an ECG was performed at week 5, before increasing the dose to 300 mg/day, and at week 7 for patients continuing to receive this dose after study completion.
Efficacy assessments were made with the Montgomery-Åsberg Depression Rating Scale (16) and the Clinical Global Impression Scale (CGI) (17). The Montgomery-Åsberg Depression Rating Scale was administered at screening or baseline and weeks 3, 5, and 7 or at early withdrawal, and the CGI was administered at weeks 2, 3, 5, and 7.
A serum sample for human norepinephrine transporter and human 5-HT transporter uptake assays was collected at the predose or baseline visit and then at weeks 2, 3, 4, 5, 6, and 7. The samples were coded, and individuals performing the uptake assays were blinded to all treatment variables, although all samples from a given individual were identified as such. An additional blood sample was obtained to determine paroxetine or desipramine serum concentrations by means of high-performance liquid chromatography. The limit of detection was 10 ng/ml for paroxetine and 25 ng/ml for desipramine. All samples were frozen at –20°C until assayed.
Selection of Subjects
Fifty-two patients (aged 18–57 years) were enrolled in this study. Entrance criteria included a primary diagnosis of major depressive disorder (DSM-IV 296.2 or 296.3) and a score ≥20 on the Montgomery-Åsberg Depression Rating Scale at screening and baseline. Exclusion criteria included a diagnosis of any axis I disorder other than major depressive disorder as the primary or predominant psychiatric diagnosis within 6 months before the screening visit; previous unresponsiveness to treatment with, hypersensitivity to, or intolerance of paroxetine or desipramine; current use of other antidepressants (tricyclic antidepressants, SSRIs, 5-HT/norepinephrine reuptake inhibitors), lithium, herbal preparations or supplements that affect the central nervous system, or oral antipsychotic medications 14 days before screening; use of MAOIs or fluoxetine within 4 weeks before screening; use of depot antipsychotic medications within 12 weeks before screening; requirement for concomitant psychotherapy; significant abnormal laboratory test or ECG findings at screening; fulfillment of DSM-IV criteria for substance abuse or dependence within 6 months or having tested positive for illicit drug use at screening; pregnancy, lactation, or lack of use of a clinically accepted method of contraception; posing a current suicidal or homicidal risk; having a history of cardiovascular disease, urinary retention, glaucoma, thyroid disease, or seizure disorder; and being judged by the investigator to have any other clinically significant condition that would preclude the administration of paroxetine or desipramine.
The principles of informed consent in the current Declaration of Helsinki (Protocol Appendix A) were implemented before initiation of the protocol. After a complete description of the study to subjects, written informed consent was obtained by using a form approved by the institutional review board at each location. This protocol was also approved by the Emory University Human Investigation Committee.
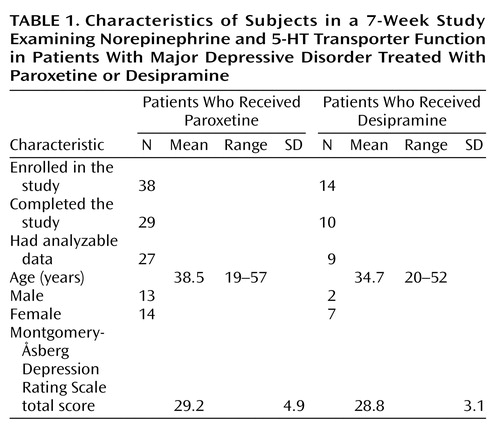
Demographic characteristics of the study participants are shown in Table 1. Overall, 52 patients were enrolled in the study. Of the 38 patients assigned to the paroxetine group, 29 completed the study and 27 had data that were analyzed. Reasons for not being included in the final analysis were lack of completion of the study due to adverse effects (N=5), lack of efficacy (N=1), or other reasons (N=5) (e.g., not drug free at baseline). Of the 14 patients assigned to the desipramine group, 10 completed the study and 9 had data that were analyzed. Reasons for not being included in the final analysis for the desipramine group were similar to those for the paroxetine group.
Uptake Assays
[3H]Norepinephrine uptake and [3H]5-HT uptake were determined as previously described (11) with modifications for serum as the assay medium. To determine standard uptake values, normal human serum (Atlanta Biologicals, Atlanta) was spiked with increasing concentrations of paroxetine or desipramine and incubated at 37°C for at least 1 hour before assay to allow free and protein-bound drug to equilibrate. In each experiment, all concentrations of a given drug were assayed in triplicate in a single 24-well plate for each cell line. The patients’ serum samples were warmed to 37°C, and samples from a single individual were assayed in triplicate in a single 24-well plate for each cell line. A triplicate sample of normal human serum was also assayed in each plate to serve as an interindividual control.
Transporter uptake was assayed by aspirating the cell culture media and washing the plated cells with 0.5 ml of phosphate-buffered saline (pH=7.2) (Sigma, St. Louis, Mo.). Three hundred microliters of each serum sample was loaded into triplicate wells and preincubated for exactly 5 minutes at 37°C, after which 10 ml of [3H]norepinephrine or [3H]5-HT (40 nM final concentration; Amersham, Piscataway, N.J.) were added and the sample was incubated at 37°C for an additional 5 minutes. The assay was terminated by aspirating the serum and washing the cells with 1.0 ml of phosphate-buffered saline. The cells were lysed with 500 ml of 0.1 M sodium hydroxide, and 450 ml was transferred to liquid scintillation vials. The [3H]norepinephrine or [3H]5-HT uptake content of the cells was quantified on a scintillation counter at 50% efficiency.
Paroxetine hydrogen chloride hemihydrate was donated by SmithKline Beecham (Worthington, West Sussex, U.K.). Desipramine, norepinephrine, and 5-HT were purchased from Sigma (St. Louis).
Data Analysis
Data points for the standard uptake curve were the mean of three separate assays of each concentration of either paroxetine or desipramine for each cell line. Individual points from patient data were obtained by expressing the average of counted triplicate values from a given sample as the percent control of the triplicate average of the predrug/baseline sample for that individual. Standard and patient curves were generated with the nonlinear, curve-fitting program PRISM (GraphPad, San Diego, Calif.) by using a one-site competition curve to describe drug-receptor interactions (18). Because drug serum levels vary widely among patients, competition curves were generated by treating the data points from all analyzed individuals in each treatment group as individual data points rather than generating a curve for each patient and then averaging all of these curves. Means are presented with standard deviations. Statistical analysis of uptake at different doses was conducted with SAS (SAS Institute, Cary, N.C.).
Results
Standard Curves
Standard curves were generated to determine the relationship between transporter inhibition and serum drug levels in the modified uptake assay (Figure 1). In normal human serum spiked and equilibrated with increasing amounts of paroxetine, increasing amounts of paroxetine were associated with a concentration-dependent decrease in norepinephrine and 5-HT uptake. At a serum paroxetine concentration of 100 ng/ml, which is often attained in therapeutic dosing, there was a 15% decrease from baseline in norepinephrine uptake and a 90% decrease in 5-HT uptake.
Normal human serum spiked and equilibrated with increasing amounts of desipramine maximally decreased norepinephrine uptake at a serum desipramine concentration of 25 ng/ml. Higher desipramine concentrations resulted in a concentration-dependent decrease in 5-HT uptake.
Clinical Data—Paroxetine
The dose of paroxetine, corresponding to weekly time points, was compared to the mean amounts of monoamine uptake and the mean total Montgomery-Åsberg Depression Rating Scale score (Figure 2). After 2 weeks of treatment with 20 mg/day of paroxetine (week 3), norepinephrine uptake and 5-HT uptake were 93.3% (SD=18.8%) and 35.7% (SD=16.4%) of control, respectively; the patients’ mean serum concentration of paroxetine was 34.9 ng/ml (SD=21.8); and their mean score on the Montgomery-Åsberg Depression Rating Scale score was 16.7 (SD=8.2). After 2 subsequent weeks of treatment with 40 mg/day of paroxetine (week 5), norepinephrine uptake and 5-HT uptake were 67.8% (SD=17.7%) and 17.1% (SD=8.0%) of control, respectively, indicating prominent inhibition of the norepinephrine transporter; the patients’ mean serum paroxetine level was 101 ng/ml (SD=52.7); and their mean Montgomery-Åsberg Depression Rating Scale score was 11.1 (SD=6.0). This dose-dependency continued after the final 2 weeks of treatment with 60 mg/day of paroxetine (week 7), when norepinephrine uptake and 5-HT uptake decreased to 58.5% (SD=16.5%) and 10.9% (SD=4.8%) of control, respectively; patients’ mean serum paroxetine level was 183 ng/ml (SD=88.3); and their mean Montgomery-Åsberg Depression Rating Scale score was 8.0 (SD=7.9).
As a given dose of paroxetine results in a wide range of serum paroxetine concentrations in the human population, we also compared serum paroxetine concentrations to monoamine uptake inhibition (Figure 3). These competition curves show that, at a serum concentration of paroxetine of 100 ng/ml, norepinephrine and 5-HT uptake were 73% and 15% of control, respectively. At a serum concentration of 200 ng/ml, norepinephrine and 5-HT uptake were 43% and 10% of control, respectively. These data indicate that the degree of norepinephrine transporter inhibition increased as serum paroxetine concentration increased, consistent with receptor binding theory.
Clinical Data—Desipramine
The serum concentration of desipramine corresponding to weekly time points was compared to the mean amount of monoamine uptake and the mean total Montgomery-Åsberg Depression Rating Scale score (Figure 4). After 2 weeks of treatment with 100 mg/day of desipramine (week 3), norepinephrine uptake and 5-HT uptake were 15.0% (SD=9.4%) and 81.8% (SD=9.8%) of control, respectively; the patients’ mean serum concentration of desipramine was 66.4 ng/ml (SD=60.6); and their mean Montgomery-Åsberg Depression Rating Scale score was 15.4 (SD=7.2). After 2 subsequent weeks of treatment with 200 mg/day of desipramine (week 5), norepinephrine uptake was maximally inhibited to 7.0% (SD=4.6%) of control and 5-HT uptake to 74.9% (SD=18.4%) of control; the patients’ mean serum concentration of desipramine was 178.4 ng/ml (SD=141.1); and their mean Montgomery-Åsberg Depression Rating Scale score was 13.6 (SD=5.6). Uptake of 5-HT continued to decrease at a dose of 300 mg/day (week 7) to 63.4% (SD=9.5%) of control. At week 7, the patients’ mean serum desipraime concentration was 279.8 ng/ml (SD=215.3), and their mean Montgomery-Åsberg Depression Rating Scale score was 10.8 (SD=9.5).
Serum desipramine concentrations were correlated with transporter inhibition (Figure 3). Norepinephrine uptake was near maximal inhibition of 15% of control at a serum desipramine concentration of 100 ng/ml, whereas 5-HT uptake was 82% of control. At a serum concentration of 500 ng/ml, 5-HT uptake decreased further to 49% of control.
Discussion
Previous in vitro binding studies indicated that paroxetine possesses moderate affinity for the human norepinephrine transporter in addition to a very high affinity for the human 5-HT transporter (11–14); however, this moderate affinity did not necessarily imply that norepinephrine transporter inhibition would occur in vivo. When rats were treated with paroxetine to a steady-state serum concentration, the norepinephrine transporter was inhibited 20% at serum concentrations of more than 100 ng/ml and 34% at serum concentrations of more than 500 ng/ml (15). These data suggested that at the doses higher than 100 ng/ml generally required to achieve serum concentrations, paroxetine would in fact act as a dual uptake inhibitor, or a 5-HT/norepinephrine reuptake inhibitor, in depressed patients. In the current study, which used a novel ex vivo assay in which patients’ serum samples were directly applied to norepinephrine transporter or 5-HT transporter transfected cells, the results clearly reveal that paroxetine inhibits the norepinephrine transporter at a level of 27% of control at a serum concentration of 100 ng/ml and more than 50% at the highest serum concentrations obtained during clinically relevant dosing (Figure 3). These results suggest that selectivity data from in vitro receptor binding assays may not always predict in vivo actions. Therefore, drugs such as paroxetine and desipramine, which have been thought to be selective in vivo, i.e., an SSRI or a noradrenergic reuptake inhibitor, respectively, clearly are not.
The recommended dose of paroxetine for treatment of depression ranges from 20 to 50 mg/day (Physicians’ Desk Reference, 1996), though doses of up to 80 mg/day have been well tolerated (19). It is well documented that a given dose of paroxetine results in a wide range of steady-state serum paroxetine concentrations among individuals (20). However, generalizations can be made, particularly in regard to the 100 ng/ml serum concentration, for which the current data indicate a 27% inhibition of the norepinephrine transporter. Doses of 20 mg/day consistently result in serum concentrations of less than 100 ng/ml, often close to the 35 ng/ml average observed in the current study (21–23). At 30 mg/day, average serum concentrations range from 55 to 90 ng/ml (21–25), with one study reporting a mean of 127 ng/ml (26). Certainly at this dose, some patients have serum paroxetine concentrations that are greater than 100 ng/ml. For these patients, paroxetine would antagonize both the norepinephrine transporter and the 5-HT transporter. At doses of 40 mg/day and higher, however, serum paroxetine concentrations are frequently higher than 100 ng/ml (21–23, 26); at those doses, paroxetine may be functioning as a 5-HT/norepinephrine reuptake inhibitor in many if not most patients.
One might argue that the transfected cells in our assay were not exposed to concentrations that mirror those actually seen in the synapses of noradrenergic and serotonergic axon terminals in the brain. Admittedly, we did not determine if free serum paroxetine concentrations were equivalent to those in the CNS. To determine CNS drug concentrations, one would have to measure CSF concentrations after lumbar puncture, an invasive procedure that was not part of the current protocol. However, several studies have shown a strong correlation between CSF and serum concentrations of several classes of psychopharmacological agents. For example, a nearly perfect correlation between CSF concentrations and predicted free-serum concentrations exists for imipramine and desipramine (27). There is also a significant correlation for the typical antipsychotic haloperidol (28, 29), other tricyclic antidepressants (30, 31), and SSRI antidepressants such as fluoxetine (32) and paroxetine (33). Paroxetine has been reported to be 95% protein bound at therapeutic serum concentrations (Physicians’ Desk Reference, 1996; 21), leaving 5% free and able to cross the blood-brain barrier (Figure 5). Lundmark et al. (33) obtained an average CSF and serum paroxetine concentration of 2.5% that approximated the estimated free concentration, which was not directly measured in the corresponding serum samples. Nonetheless, because human serum was the vehicle in our assay, the free paroxetine in the serum samples would be expected to reflect the paroxetine concentration in the patients’ CSF. It would follow, therefore, that paroxetine concentrations in the brain would be sufficient to inhibit norepinephrine transporter at the same free concentrations used in this study.
If paroxetine acts as a 5-HT/norepinephrine reuptake inhibitor at higher concentrations, one would expect it to have properties similar to those of the 5-HT/norepinephrine reuptake inhibitor venlafaxine. In vitro data have indicated that venlafaxine inhibits 5-HT uptake at low doses and both norepinephrine and 5-HT uptake at higher doses (9, 34). Similarly, the standard-curve data presented in Figure 1 and previous animal data (15) indicated that paroxetine inhibits 5-HT at serum levels that result from a low dose (i.e., 20 mg/day) and both norepinephrine and 5-HT at serum levels that result from a higher dose (i.e., 40 mg/day).
Recent indirect evidence in healthy normal human volunteers has supported the view that venlafaxine has dual uptake blocking properties in vivo (35), although the magnitude of norepinephrine transporter inhibition remains obscure, and this phenomenon has not been studied in depressed subjects. In fact, several groups have claimed that venlafaxine is superior to the SSRIs, including paroxetine, in treatment of severe and refractory depression (36, 37). The current data presented in Figure 2 and Figure 3 support the view that paroxetine also possesses dual uptake blocking properties in vivo in depressed patients. Moreover, these results may provide a rationale for the finding that a 40-mg/day dose of paroxetine to be more efficacious in preventing depressive recurrences than a 20 mg/day dose in patients previously treated effectively with a 40 mg/day dose (38). In addition, there is evidence that paroxetine is an effective treatment for refractory depression at the higher doses of 30–40 mg/day at which norepinephrine transporter inhibition is most likely to be occurring (39).
On the surface, direct comparisons of venlafaxine and paroxetine appear to indicate that venlafaxine is more efficacious than paroxetine (6, 36, 37). However, in these studies low doses of paroxetine were often compared to high doses of venlafaxine, and serum paroxetine concentrations were not reported when doses of 30–40 mg/day were used. In effect, therefore, these studies have compared only the SSRI properties of low doses or probable low serum paroxetine levels to the 5-HT/norepinephrine reuptake inhibitor properties of venlafaxine at high doses. A true comparison of the 5-HT/norepinephrine reuptake inhibitor properties of these drugs would require comparing the drugs at doses that resulted in dual uptake inhibition.
Within the range of doses utilized in this study, desipramine was also found to be a dual uptake inhibitor. Desipramine has a nearly 100-fold greater selectivity for the norepinephrine transporter than for the 5-HT transporter in vitro (11), and the potency of desipramine as an inhibitor of norepinephrine transporter often leads to desipramine’s being classified as a “pure” noradrenergic reuptake inhibitor. However, desipramine also possesses moderate potency for the 5-HT transporter in vitro. Indeed, analogous to the current findings, desipramine, like other 5-HT transporter inhibitors, has been found to decrease whole blood 5-HT concentrations in humans by inhibition of the platelet 5-HT transporter (40).
There is growing evidence that using either a combination of medications that inhibit the norepinephrine transporter and the 5-HT transporter or a single medication that inhibits both transporters may be more effective in addressing severe and treatment-resistant depression and may shorten the onset of therapeutic action. Although venlafaxine has been singularly promoted as a 5-HT/norepinephrine reuptake inhibitor, several antidepressants are in fact dual uptake inhibitors and perhaps 5-HT/norepinephrine reuptake inhibitors. The results of the current study demonstrate that paroxetine can act as a 5-HT/norepinephrine reuptake inhibitor at higher therapeutic serum concentrations. The physiological and clinical significance of the partial norepinephrine transporter inhibition in the presence of nearly complete 5-HT transporter inhibition is unclear. However, a large amount of norepinephrine transporter inhibition may not be needed when combined with nearly complete 5-HT transporter inhibition. For example, the only estimation of norepinephrine transporter occupancy during high-dose venlafaxine treatment showed only partial inhibition of the norepinephrine transporter, as assessed with measures of blood pressure in response to challenge with the false transmitter tyramine (35).
In the absence of direct measures of norepinephrine transporter occupancy in vivo in depressed patients (e.g., use of ligands in positron emission tomography or single photon emission computed tomography), this novel ex vivo uptake assay may accurately predict CNS transporter occupancy. Our results suggest that paroxetine, a potent SSRI, inhibits the norepinephrine transporter and is a 5-HT/norepinephrine reuptake inhibitor at high therapeutic serum levels. Thus, classifications of drugs based exclusively on their binding profile in vitro may need to be reconsidered. Finally, whether added norepinephrine transporter inhibition results in increased efficacy or decreased latency to efficacy remains to be conclusively determined.
 |
Presented at the World Federation of Biological Psychiatry, Berlin, July 1–6, 2001 and the 31st Annual Meeting of the Society for Neuroscience, San Diego, Nov. 10–15, 2001. Received Sept. 4, 2001; revision received April 10, 2002; accepted April 18, 2002. From the Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine. Address reprint requests to Dr. Nemeroff, Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine, 1639 Pierce Dr., Suite 4000, WMB Bldg., Atlanta, GA 30322; [email protected] (e-mail). Supported by an unrestricted grant from GlaxoSmithKline. The authors thank Susan Plott and David Knight for technical assistance.

Figure 1. Relationship Between Norepinephrine and 5-HT Uptake and Concentrations of Paroxetine and Desipramine in Normal Human Seruma
aEach point represents the mean of three separate assays of each concentration of either paroxetine or desipramine in normal human serum. A one-site competition curve used to describe drug-transporter interactions resulted in a goodness-of-fit value of R2=0.99 for every condition. Drug concentrations corresponding to the log units are shown above each curve. The limit of detection for serum paroxetine and desipramine concentrations is 10 ng/ml and 25 ng/ml, respectively.

Figure 2. Norepinephrine and 5-HT Uptake, Serum Paroxetine Levels, and Montgomery-Åsberg Depression Rating Scale Scores in Patients With Major Depressive Disorder (N=27) During 7 Weeks of Treatment With Paroxetinea
aSignificant inhibition of norepinephrine and 5-HT uptake at weeks 3, 5, and 7. For norepinephrine: F=64.94, df=2, 26, p<0.0001, single-factor analysis with repeated measures by dose and week; significant post hoc differences between weeks 3 and 5 (p<0.001, Tukey), weeks 3 and 7 (p<0.001, Tukey), and weeks 5 and 7 (p=0.01, Tukey). For 5-HT: F=50.9, df=2, 26, p<0.0001, single-factor analysis with repeated measures by dose and week; significant post hoc differences between weeks 3 and 5 (p<0.001, Tukey), weeks 3 and 7 (p<0.001, Tukey), and weeks 5 and 7 (p<0.001, Tukey).

Figure 3. Relationship Between Norepinephrine and 5-HT Uptake and Serum Concentrations of Paroxetine and Desipramine in Patients With Major Depressive Disordera
aCurves were generated from data for 27 patients who received paroxetine and nine patients who received desipraimine. Classic one-site competition curves used to describe drug-transporter interactions resulted in goodness-of-fit values of R2=0.51 for norepinephrine uptake in patients taking paroxetine, R2=0.96 for 5-HT uptake in patients taking paroxetine, R2=0.99 for norepinephrine uptake in patients taking desipramine, R2=0.59 for 5-HT uptake in patients taking desipramine. Drug concentrations corresponding to the log units are shown above each curve. In the panel depicting norepinephrine uptake for paroxetine, six data points ranging from 125% to 150% of control were not shown to assist in visual comparison among the panels. These data points were used, however, to generate the competition curve.

Figure 4. Norepinephrine and 5-HT Uptake, Serum Desipramine Levels, and Montgomery-Åsberg Depression Rating Scale Scores in Patients With Major Depressive Disorder (N=9) During 7 Weeks of Treatment With Desipraminea
aSignificant inhibition of norepinephrine and 5-HT uptake at weeks 3, 5, and 7. For norepinephrine: F=5.71, df=2, 8, p<0.05, single-factor analysis with repeated measures by dose and week; significant post hoc differences between weeks 3 and 5 (p<0.05, Tukey), weeks 3 and 7 (p<0.05, Tukey), and weeks 5 and 7 (p=0.77, Tukey). For 5-HT: F=19.49, df=2, 8, p<0.001, single-factor analysis with repeated measures by dose and week; significant post hoc differences between weeks 3 and 5 (p=0.26, Tukey), weeks 3 and 7 (p<0.05, Tukey), and weeks 5 and 7 (p<0.01, Tukey).

Figure 5. Relationship Between Free Serum Drug Concentration and CNS Extracellular Water Drug Concentrationa
aMany drugs, including paroxetine and desipramine, are weak bases that are highly protein bound in serum in their charged form. An equilibrium exists between the charged and neutral state, in which the free uncharged drug is able to pass through the blood-brain barrier into the central nervous system (CNS). An equilibrium also exists in the physiological pH of the extracellular fluid within the CNS, allowing the drug to remain in the charged state, in which it is able to exert clinical effects. Theoretically, free drug concentration in serum should be similar to that in CSF. Illustration by Jennifer Gentry.
1. Owens MJ, Mulchahey JJ, Stout SC, Plotsky PM: Molecular and neurobiological mechanisms in the treatment of psychiatric disorders, in Psychiatry, vol 1. Edited by Tasman A, Kay J, Lieberman JA. Philadelphia, WB Saunders, 1997, pp 210-257Google Scholar
2. Nelson JC: A review of the efficacy of serotonergic and noradrenergic reuptake inhibitors for treatment of major depression. Biol Psychiatry 1999; 46:1301-1308Crossref, Medline, Google Scholar
3. Ressler KJ, Nemeroff CB: Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety 2000; 12(suppl 1):2-19Google Scholar
4. Steffens DC, Krishnan KRR, Helms MJ: Are SSRIs better than TCAs? comparison of SSRIs and TCAs: a meta-analysis. Depress Anxiety 1997; 6:10-18Crossref, Medline, Google Scholar
5. Nelson JC: Synergistic benefits of serotonin and noradrenaline reuptake inhibition. Depress Anxiety 1998; 7(suppl 1):5-6Google Scholar
6. Thase ME, Entsuah AR, Rudolph RL: Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001; 178:234-241Crossref, Medline, Google Scholar
7. Nelson JC, Mazure CM, Bowers Jr. MB, Jatlow PI: A preliminary, open study of the combination of fluoxetine and desipramine for rapid treatment of major depression. Arch Gen Psychiatry 1991; 48:303-307Crossref, Medline, Google Scholar
8. Seth R, Jennings AL, Bindman J, Phillips J, Bergmann K: Combination treatment with noradrenalin and serotonin reuptake inhibitors in resistant depression. Br J Psychiatry 1992; 161:562-565Crossref, Medline, Google Scholar
9. Muth EA, Haskins JT, Moyer JA, Husbands GE, Nielsen ST, Sigg EB: Antidepressant biochemical profile of the novel bicyclic compound Wy-45,030, an ethyl cyclohexanol derivative. Biochem Pharmacol 1986; 35:4493-4497Crossref, Medline, Google Scholar
10. Potter WZ, Manji HK, Rudorfer MV: Tricyclics and tetracyclics, in The American Psychiatric Press Textbook of Psychopharmacology. Edited by Schatzberg AF, Nemeroff CB. Washington, DC, American Psychiatric Press, 1998, pp 199-218Google Scholar
11. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB: Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther 1997; 283:1305-1322Medline, Google Scholar
12. Tatsumi M, Groshan K, Blakely RD, Richelson E: Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 1997; 340:249-258Crossref, Medline, Google Scholar
13. Beique J-C, Lavoie N, de Montigny C, Debonnel G: Affinities of venlafaxine and various reuptake inhibitors for the serotonin and norepinephrine transporters. Eur J Pharmacol 1998; 349:129-132Crossref, Medline, Google Scholar
14. Owens MJ, Knight DL, Nemeroff CB: Second generation SSRIs: human monoamine transporter binding profile of S-citalopram and R-fluoxetine. Biol Psychiatry 2001; 50:345-350Crossref, Medline, Google Scholar
15. Owens MJ, Knight DL, Nemeroff CB: Paroxetine binding to the rat norepinephrine transporter in vivo. Biol Psychiatry 2000; 47:842-845Crossref, Medline, Google Scholar
16. Montgomery SA, Åsberg M: A new depression scale designed to be sensitive to change. Br J Psychiatry 1979; 134:382-389Crossref, Medline, Google Scholar
17. Guy W (ed): ECDEU Assessment Manual for Psychopharmacology: Publication ADM 76-338. Washington, DC, US Department of Health, Education, and Welfare, 1976, pp 218-222Google Scholar
18. Kenakin T: Pharmacological Analysis of Drug-Receptor Interaction. New York, Lippincott-Raven, 1997Google Scholar
19. Dunner DL, Dunbar GC: Optimal dose regimen for paroxetine. J Clin Psychiatry 1992; 53(suppl 2):21-26Google Scholar
20. Gunasekara NS, Noble S, Benfield P: Paroxetine: an update of its pharmacology and therapeutic use in depression and a review of its use in other disorders. Drugs 1998; 55:85-120Crossref, Medline, Google Scholar
21. Kaye CM, Haddock RE, Langley PF, Mellows G, Tasker TCG, Zussman BD, Greb WH: A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr Scand Suppl 1989; 350:60-75Crossref, Medline, Google Scholar
22. DuBoff EA: Long-term treatment of major depressive disorder with paroxetine. J Clin Psychopharmacol 1993; 13(suppl 2):28S-33SGoogle Scholar
23. Lundmark J, Bengtsson F, Nordin C, Reis M, Walinder J: Therapeutic drug monitoring of selective serotonin reuptake inhibitors influences clinical dosing strategies and reduces drug costs in depressed elderly patients. Acta Psychiatr Scand 2000; 101:354-359Crossref, Medline, Google Scholar
24. Tasker TCG, Kaye CM, Zussman BD, Link CGG: Paroxetine plasma levels: lack of correlation with efficacy or adverse events. Acta Psychiatr Scand Suppl 1989; 350:152-155Crossref, Medline, Google Scholar
25. Danish University Antidepressant Group: Paroxetine: a selective serotonin reuptake inhibitor showing better tolerance, but weaker antidepressant effect than clomipramine in a controlled multicenter study. J Affect Disord 1990; 18:289-299Crossref, Medline, Google Scholar
26. Sindrup SH, Brosen K, Gram LF: Pharmacokinetics of the selective serotonin reuptake inhibitor paroxetine: nonlinearity and relation to the sparteine oxidation polymorphism. Clin Pharmacol Ther 1992; 51:288-295Crossref, Medline, Google Scholar
27. Muscettola G, Goodwin FK, Potter WZ, Claeys MM, Markey SP: Imipramine and desipramine in plasma and spinal fluid: relationship to clinical response and serotonin metabolism. Arch Gen Psychiatry 1978; 35:621-625Crossref, Medline, Google Scholar
28. Forsman A, Ohman R: Studies on serum protein binding of haloperidol. Curr Ther Res Clin Exp 1977; 21:245-255Medline, Google Scholar
29. Linkowski P, Hubain P, von Frenckell R, Mendlewicz J: Haloperidol plasma levels and clinical response in paranoid schizophrenics. Eur Arch Psychiatry Neurol Sci 1984; 234:231-236Crossref, Medline, Google Scholar
30. Hanin I, Koslow SH, Kocsis JH, Bowden CL, Brunswick D, Frazer A, Carl J, Robins E: Cerebrospinal fluid levels of amitriptyline, nortriptyline, imipramine and desmethylimipramine. J Affect Disord 1985; 9:69-78Crossref, Medline, Google Scholar
31. Potter WZ, Scheinin M, Golden RN, Rudorfer MV, Wowdry RW, Calil HM, Ross RJ, Linnoila M: Selective antidepressants and cerebrospinal fluid: lack of specificity on norepinephrine and serotonin metabolites. Arch Gen Psychiatry 1985; 42:1171-1177Crossref, Medline, Google Scholar
32. Martensson B, Nyberg S, Toresson G, Brodin E, Bertilsson L: Fluoxetine treatment of depression. Acta Psychiatr Scand 1989; 79:586-596Crossref, Medline, Google Scholar
33. Lundmark J, Walinder J, Alling C, Manniche PM, Dalgaard L: The effect of paroxetine on cerebrospinal fluid concentrations of neurotransmitter metabolites in depressed patients. Eur Neuropsychopharmacol 1994; 4:1-6Crossref, Medline, Google Scholar
34. Andrews JM, Ninan PT, Nemeroff CB: Venlafaxine: a novel antidepressant that has a dual mechanism of action. Depression 1996; 4:48-56Crossref, Medline, Google Scholar
35. Harvey AT, Rudolph RL, Preskorn SH: Evidence of the dual mechanisms of action of venlafaxine. Arch Gen Psychiatry 2000; 57:503-509Crossref, Medline, Google Scholar
36. Poirier M-F, Boyer P: Venlafaxine and paroxetine in treatment-resistant depression. Br J Psychiatry 1999; 175:12-16Crossref, Medline, Google Scholar
37. Ballus C, Quiros G, De Flores T, de la Torre J, Palao D, Rojo L, Gutierrez M, Casais L, Riesgo Y: The efficacy and tolerability of venlafaxine and paroxetine in outpatients with depressive disorder or dysthymia. Int Clin Psychopharmacol 2000; 15:43-48Crossref, Medline, Google Scholar
38. Franchini L, Gasperini M, Perez J, Smeraldi E, Zanardi R: Dose-response efficacy of paroxetine in preventing depressive recurrences: a randomized, double-blind study. J Clin Psychiatry 1998; 59:229-232Crossref, Medline, Google Scholar
39. Tyrer P, Marsden CA, Casey P, Seivewright N: Clinical efficacy of paroxetine in resistant depression. J Psychopharmacol 1987; 1:251-257Crossref, Medline, Google Scholar
40. Javors MA, Houston JP, Tekell JL, Brannan SK, Frazer A: Reduction of platelet serotonin content in depressed patients treated with either paroxetine or desipramine. Int J Neuropsychopharmacol 2000; 3:229-235Crossref, Medline, Google Scholar

