
Synaptic Mechanisms Underlying Rapid Antidepressant Action of Ketamine
Abstract
Recent clinical studies have demonstrated that a single subpsychotomimetic dose of ketamine, an ionotropic glutamatergic N-methyl-d-aspartate (NMDA) receptor antagonist, produces a rapid antidepressant response in patients with major depressive disorder, with effects lasting up to 2 weeks. Despite enthusiasm about this unexpected efficacy of ketamine, its widespread use as a fast-acting antidepressant in routine clinical settings is curtailed by its abuse potential as well as possible psychotomimetic effects. However, the ability of ketamine to produce a rapid and long-lasting antidepressant response in patients with depression provides a unique opportunity for investigation of mechanisms that mediate these clinically relevant behavioral effects. From a mechanistic perspective, it is easy to imagine how activation of NMDA receptors may trigger cellular and behavioral responses; it is relatively more difficult, however, to envision how transient blockade of one of the key pathways for neuronal communication produces a persistent beneficial effect. The authors discuss recent work linking ketamine’s mechanism of action to homeostatic synaptic plasticity processes activated after suppression of NMDA-mediated glutamatergic neurotransmission. They focus on their recent work demonstrating that ketamine-mediated blockade of NMDA receptors at rest deactivates eukaryotic elongation factor 2 (eEF2) kinase, resulting in reduced eEF2 phosphorylation and desuppression of rapid dendritic protein translation, including BDNF (brain-derived neurotrophic factor), which then contributes to synaptic plasticity mechanisms that mediate long-term effects of the drug. The authors also explore possible molecular strategies to target spontaneous neurotransmitter release selectively to help uncover novel presynaptic avenues for the development of fast-acting antidepressants and possibly psychoactive compounds with effectiveness against other neuropsychiatric disorders.
Estimates from the National Institute of Mental Health indicate that the prevalence of major depressive disorder is 6.4% of the U.S. adult population. A significant number of patients with depression do not respond to currently available medications, such as selective serotonin reuptake inhibitors, and even in cases of successful treatment, these compounds typically take weeks or months to trigger an antidepressant response. This delay in onset is a major drawback to current antidepressant therapies, leaving a crucial need for the development of faster-acting antidepressants, especially in patients at risk of suicide. Recent clinical studies have consistently demonstrated that a single subpsychotomimetic dose of ketamine, an ionotropic glutamatergic N-methyl-d-aspartate (NMDA) receptor antagonist, produces a rapid antidepressant response in patients suffering from major depression. Treatment-resistant depressed patients reported alleviation of core symptoms of major depression within hours of a single low-dose intravenous infusion of ketamine, with effects lasting up to 2 weeks (1–3). Despite enthusiasm about the unexpected efficacy of ketamine, its widespread use as a fast-acting antidepressant in routine clinical settings is curtailed by its abuse potential as well as possible psychotomimetic effects. However, the ability of ketamine to produce a fast-acting and long-lasting antidepressant response in depressed patients provides a unique opportunity for preclinical investigation of cellular mechanisms that mediate these clinically relevant behavioral effects.
NMDA Receptor Blockade and Neuronal Activity
The antidepressant effects of ketamine are reportedly sustained for approximately 1–2 weeks after acute administration, indicating a key role of neuronal plasticities in the mediation of the drug’s behavioral effects. Accordingly, in animal models, acute ketamine administration has been shown to activate several downstream signaling cascades that include mammalian target of rapamycin (mTor) and glycogen synthase kinase-3 (GSK3), which have been implicated in plasticity mechanisms as well as the pathophysiology of neuropsychiatric disorders (4, 5). However, these signal transduction cascades can also be activated indirectly through increases in synaptic efficacy and subsequent increases in neuronal activity. Indeed, earlier work has shown that increases in synaptic efficacy, as seen after long-term potentiation of synaptic inputs, can augment the ability of individual synapses to elicit neuronal action potential firing (6). Therefore, an increase in synaptic efficacy may lead to an overall increase in neuronal excitability and activate multiple downstream signaling cascades that include mTor and GSK3. In view of these findings, studies that have been conducted on downstream targets of ketamine action leave open the question as to how NMDA receptor blockade by ketamine, which would be expected to decrease synaptic transmission and long-term potentiation, instead leads to coordinated increases in postsynaptic signaling and synaptic efficacy, which in turn may have profound influences on synaptic network activity patterns.
The basic property of ketamine as a use-dependent blocker of NMDA receptor-mediated neurotransmission suggests that the cellular and behavioral effects of this compound are triggered by suppression of a tonically active form of glutamatergic signaling in the brain. A prevailing hypothesis posits that ketamine-induced suppression of tonic NMDA receptor-mediated glutamatergic input onto GABA-ergic interneurons leads to a decrease in overall inhibition, also called disinhibition, tilting the balance of synaptic transmission toward excitation. The model envisions a purely electrical effect of NMDA receptor blockade on subsequent signaling and overall neuronal activity (7). Yet, it is important to note that tonic glutamatergic transmission is also expected to activate ionotropic glutamatergic 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors, as most mature synapses contain both AMPA and NMDA receptor populations (8, 9).
Spontaneous and Evoked Neurotransmission
Studies in the past two decades have dissected the neurotransmitter release process into molecularly and functionally distinct components. These studies have given significant attention to the mechanisms underlying the process of rapid neurotransmitter release in response to fast neuronal spiking activity. However, these molecular and functional studies have also uncovered clear mechanistic distinctions between rapid release of a neurotransmitter that is coupled to neuronal activity, which is referred to as evoked neurotransmission, and neurotransmitter release that occurs independently of neuronal activity, which is referred to as spontaneous neurotransmission.
Evoked neurotransmission is described as presynaptic action potential firing-driven neurotransmitter release that then acts on postsynaptic receptors to mediate specific effects on intracellular signaling cascades. There are two types of evoked neurotransmission: synchronous release of neurotransmitters, which is tightly coupled to nervous activity, and asynchronous release of neurotransmitters, which is loosely coupled to nervous activity. Evoked neurotransmission is a well-studied process that has many important functions in the CNS, including in learning and memory (12).
The second and less commonly studied form of neurotransmission, spontaneous transmission, occurs in the CNS as a result of a low but not negligible probability that a synaptic vesicle will spontaneously fuse with the presynaptic membrane. Recent evidence has demonstrated that spontaneous neurotransmission may have important roles in the CNS, including modulating action potential firing in some neurons, maturation and stability of synaptic networks, local dendritic protein synthesis, and mediating effects on homeostatic synaptic plasticity (13). Several intriguing studies have demonstrated that when glutamate is released spontaneously in the absence of presynaptic action potentials (also referred to as “at rest”), it activates specific NMDA receptors that couple to an intracellular signaling cascade to mediate cellular and molecular processes in a manner distinct from when glutamate is released through evoked transmission (14–16). The notion that spontaneous transmission may mediate distinct cellular processes from evoked neurotransmission raises intriguing possibilities about the role of this form of neurotransmission in vivo.
Spontaneous Neurotransmission and Its Impact on Neuronal Signaling
Under at-rest conditions, glutamatergic signaling can be driven by three sources. First, increases in ambient glutamate may originate from spillover of normal evoked synaptic activity (17). In this scenario, depending on the parameters that affect glutamate diffusion, clearance, and buffering, a glutamatergic tone may last several seconds beyond the time frame of rapid evoked neurotransmission. Second, baseline glutamate may be nonsynaptic in origin and driven by reversal of various transporters that can shuttle glutamate in and out of neurons (18). These mechanisms may dominate in pathological circumstances triggered by hypoxic insults such as stroke (19) and contribute to glutamatergic excitotoxicity. Third, several recent studies suggest that spontaneous quantal neurotransmitter release can also contribute to tonic glutamatergic signaling, especially in cases in which ambient glutamate levels are estimated to be extremely low (20).
In support of a role for spontaneous glutamate release in resting glutamatergic signaling, an increasing number of studies have shown that spontaneous release events trigger biochemical signaling that leads to maturation and stability of synaptic networks, local dendritic protein synthesis, and control of postsynaptic responsiveness during homeostatic synaptic plasticity (13). Interestingly, several studies have shown specific effects of postsynaptic excitatory receptor blockade or inhibition of neurotransmitter release under resting conditions, which could not be achieved by inhibition of action potential-mediated signaling alone (21). These observations suggest that selective regulation of spontaneous glutamatergic transmission may lead to specific signaling outcomes.
Impact of NMDA Receptor Blockade on Synaptic Homeostasis
To understand how ketamine may affect synaptic processes, it is instructive to turn to a historical example of how suppression of synaptic inputs can influence plasticity. A widespread phenomenon that has puzzled neurophysiologists for over a century is the overexcitability of target membranes after denervation or disruption of their nerve input (22). The mechanistic basis for this increase in effector sensitivity remains a current question in neurophysiology. Early experiments in the neuromuscular junction showed that the increase in the sensitivity of muscle tissue to acetylcholine was due to up-regulation of acetylcholine receptors (23).
Studies have also identified more local, synapse-specific, forms of homeostatic plasticity that are triggered by blockade of postsynaptic glutamate receptors, in particular NMDA receptor-mediated transmission (16, 27) or AMPA receptor-mediated transmission (28, 29). In these experiments, NMDA receptor blockade by MK-801 or (2R)-amino-5-phosphonovaleric acid (AP5), in the absence of neuronal activity, could augment protein synthesis through dephosphorylation of eukaryotic elongation factor-2 (eEF2), a critical catalytic factor for ribosomal translocation during protein synthesis (16). Notably, in these experiments, blockade of action potentials per se by application of the voltage-gated Na+ channel blocker tetrodotoxin is not sufficient to trigger eEF2 dephosphorylation. Therefore, according to this paradigm, NMDA receptor activity at rest causes chronic activation of eEF2 kinase (eEF2K; also called CaMKIII), which phosphorylates eEF2, effectively suppressing translation. In contrast, acute NMDA receptor blockade at rest prevents eEF2 phosphorylation, thereby increasing translation of target transcripts, including AMPA receptor subunits. These studies by Sutton and colleagues provided a causal link between neurotransmission occurring at rest in the absence of action potentials and synaptic homeostasis. An important point of these studies is that spontaneous neurotransmitter release, rather than evoked neurotransmission, is a specific regulator of postsynaptic sensitivity to neurotransmitters by suppressing the dendritic protein translation machinery locally and maintaining receptor composition of synapses.
A Synaptic Mechanism for Ketamine Action
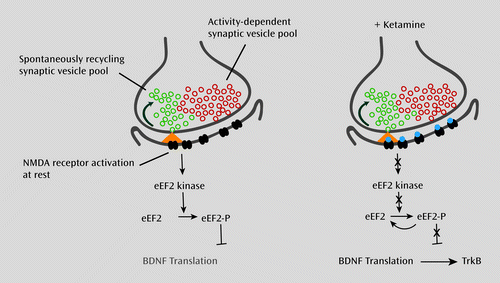
a On the left, when neurons are at rest, spontaneous glutamate release and N-methyl-d-aspartic acid (NMDA) receptor activation leads to activation of eEF2 kinase, triggering eEF2 phosphorylation and silencing of brain-derived neurotrophic factor (BDNF) translation. On the right, NMDA receptor blockade at rest, in turn, does not activate eEF2 kinase, resulting in a gradual loss of eEF2 phosphorylation and desuppression of BDNF translation, ultimately triggering TrkB (tyrosine-related kinase B) receptor signaling. Recent work suggests that synaptic vesicles giving rise to spontaneous neurotransmission (green vesicles) are in part distinct from those that give rise to action potential neurotransmitter release (red vesicles). Taken together, these observations raise the possibility that one can selectively target spontaneous neurotransmitter release to elicit rapidly acting antidepressant responses.
At this time, we do not have a clear picture of the regional specificity of the ketamine effect. In principle, the mechanisms we describe here should be present in several types of glutamatergic synapses in the CNS. However, our work so far indicates that ketamine-induced dephosphorylation of eEF2 is most detectable in hippocampus. The mechanisms that give rise to this apparent regional specificity remain to be elucidated, but there are many possibilities, among them regional differences in the rate of spontaneous glutamate release, the coupling between NMDA receptors and the eEF2 kinase signaling pathway, the availability of the BDNF transcripts, and whether all components of the dendritic protein translational machinery are present and available for fast-acting translation.
Our findings implicating the blockade of spontaneous NMDA receptors and the inhibition of eEF2 kinase to up-regulate dendritic protein translation as the trigger for ketamine’s fast-acting antidepressant action have a number of implications. First, increased baseline glutamatergic transmission may be a player in the pathophysiology of depression. This proposal is supported by a recent study that uncovered enhanced basal glutamatergic transmission in a rat model of depression (40). Moreover, several studies support the involvement of increased glutamate release in the context of depression and stress-related behaviors (41). However, it remains unclear whether there is overlap between the mechanisms that underlie the pathophysiology of depression and those involved in mediating antidepressant responses. Second, the dendritic protein translation machinery, especially processes that affect rapid translation of BDNF, appears to be a strong target for the development of novel antidepressants. Third, spontaneous glutamate release itself, when suppressed selectively, could provide a valuable target for potential antidepressant action, as discussed in the final section of this review.
Selective Regulation of Spontaneous Neurotransmitter Release
A traditional extension of the recent studies of ketamine-mediated rapid antidepressant action would entail a detailed examination of targets downstream of NMDA receptor blockade (such as inhibitors of eEF2 kinase) for development as fast-acting antidepressants. While this is a logical and worthwhile approach, it has been extremely difficult to develop kinase-selective drugs for use in patient populations. Furthermore, an NMDA receptor blocker such as ketamine mediates effects on both spontaneous and evoked transmission, although its impact on spontaneous neurotransmission appears to be necessary and sufficient to elicit the behavioral antidepressant effect. NMDA receptor activation during evoked neurotransmission is a well-characterized pathway underlying several forms of long-term synaptic plasticity, and its inhibition is known to trigger several cognitive side effects of NMDA receptor blockers; therefore, it is possible that simply developing other NMDA receptor blockers may not be enough to avoid the side effects associated with blocking evoked transmission. Indeed, it is interesting to speculate whether the adverse psychotomimetic effects observed with higher doses of ketamine may be due to a more pronounced effect on evoked neurotransmission. To circumvent these potential problems, an “upstream” strategy that selectively suppresses spontaneous neurotransmitter release and leaves evoked neurotransmission intact may present several advantages.
One possible upstream approach for selectively targeting spontaneous transmission may take advantage of the differences in presynaptic vesicles involved in the two forms of neurotransmission. A number of studies have provided evidence that presynaptic vesicle populations giving rise to spontaneous release events are distinct from those that carry out action potential-driven neurotransmission (42–44). Recent work has uncovered several molecular candidates that may be involved in this dichotomy. For instance, a noncanonical SNARE molecule, Vps10p-tail-interactor-1a (vti1a), previously shown to reside on synaptic vesicles, could only be slowly mobilized during activity compared with the more abundant vesicular SNARE synaptobrevin2 (also called VAMP2). Under resting conditions, vti1a showed robust trafficking that could be partly matched by synaptobrevin (45). Subsequent experiments demonstrated that loss of vti1a function selectively reduced spontaneous neurotransmitter release detected postsynaptically, with no effect on evoked neurotransmitter release. These results provide evidence that vti1a in its native form selectively maintains spontaneous neurotransmitter release and thus constitutes a specific marker for this form of neurotransmission. From these results, it is tempting to hypothesize that small molecules that suppress vti1a trafficking should selectively impair spontaneous release and thus mediate synaptic as well as behavioral effects that in part mimic ketamine’s antidepressant effect. One proof of principle for this approach would be to selectively delete vti1a in particular brain regions of rodents and assess the animals’ ability to respond to ketamine as a fast-acting antidepressant. The success of this type of approach would provide further support for the development of small molecules to target presynaptic targets such as vti1a for the treatment of depression.
In addition to vti1a, VAMP7 and VAMP4, which are also noncanonical synaptic vesicle-associated SNARE molecules, have been shown to specifically drive spontaneous and asynchronous forms of neurotransmission, respectively (46, 47). These examples suggest several novel presynaptic synaptic vesicle trafficking pathways that may be targeted to elicit specific alterations in neurotransmitter release and modify subsequent synaptic signal transduction events, leading to specific behavioral outcomes.
Conclusions
The recent findings on the specific presynaptic molecules that selectively regulate spontaneous release render feasible the development of screens that will identify compounds that rapidly suppress spontaneous neurotransmitter release without significantly altering evoked neurotransmission. Our prediction is that compounds that act presynaptically to selectively impair spontaneous neurotransmitter release in an acute manner should trigger a behavioral response similar to ketamine’s rapid antidepressant response but without ketamine’s side effects, which may arise from its blockade of evoked transmission. An important aspect of this strategy will be delineating acute from chronic inhibition of spontaneous neurotransmitter release, which may lead to neuronal adaptations that hinder the rapid antidepressant action. The selective suppression of spontaneous neurotransmission represents a novel and intriguing target, and if validated, this approach may uncover a novel avenue for the development of fast-acting antidepressants and possibly psychoactive compounds with effectiveness against other neuropsychiatric disorders.
1 : Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000; 47:351–354Crossref, Medline, Google Scholar
2 : Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry 2009; 66:522–526Crossref, Medline, Google Scholar
3 : A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006; 63:856–864Crossref, Medline, Google Scholar
4 : Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry 2011; 16:1068–1070Crossref, Medline, Google Scholar
5 : mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010; 329:959–964Crossref, Medline, Google Scholar
6 : Measuring the impact of probabilistic transmission on neuronal output. Neuron 1993; 10:1101–1111Crossref, Medline, Google Scholar
7 : Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 1997; 17:2921–2927Crossref, Medline, Google Scholar
8 : NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature 1989; 341:230–233Crossref, Medline, Google Scholar
9 : Spontaneous unitary synaptic activity in CA1 pyramidal neurons during early postnatal development: constant contribution of AMPA and NMDA receptors. J Neurosci 2002; 22:5552–5562Crossref, Medline, Google Scholar
10 : NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011; 475:91–95Crossref, Medline, Google Scholar
11 : Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 2008; 63:349–352Crossref, Medline, Google Scholar
12 : Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 2012; 73:990–1001Crossref, Medline, Google Scholar
13 : Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology (Bethesda) 2011; 26:45–53Crossref, Medline, Google Scholar
14 : Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci 2008; 28:10151–10166Crossref, Medline, Google Scholar
15 : NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol 2009; 101:2290–2296Crossref, Medline, Google Scholar
16 : Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 2007; 55:648–661Crossref, Medline, Google Scholar
17 : Distribution of extracellular glutamate in the neuropil of hippocampus. PLoS ONE 2011; 6:e26501Crossref, Medline, Google Scholar
18 : Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 2003; 6:743–749Crossref, Medline, Google Scholar
19 : Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000; 403:316–321Crossref, Medline, Google Scholar
20 : Extracellular glutamate concentration in hippocampal slice. J Neurosci 2007; 27:9736–9741Crossref, Medline, Google Scholar
21 : Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol 2011; 21:275–282Crossref, Medline, Google Scholar
22 : The Supersensitivity of Denervated Structures: A Law of Denervation. New York, Macmillan, 1949Google Scholar
23 : A study of supersensitivity in denervated mammalian skeletal muscle. J Physiol 1959; 147:178–193Crossref, Medline, Google Scholar
24 : Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 2004; 5:97–107Crossref, Medline, Google Scholar
25 : Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci 2005; 25:2895–2905Crossref, Medline, Google Scholar
26 : Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol 2006; 96:2127–2133Crossref, Medline, Google Scholar
27 : Dendritic protein synthesis, synaptic plasticity, and memory. Cell 2006; 127:49–58Crossref, Medline, Google Scholar
28 : Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 2010; 68:1143–1158Crossref, Medline, Google Scholar
29 : Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci USA 2010; 107:21806–21811Crossref, Medline, Google Scholar
30 : A neurotrophic model for stress-related mood disorders. Biol Psychiatry 2006; 59:1116–1127Crossref, Medline, Google Scholar
31 : Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 2001; 50:260–265Crossref, Medline, Google Scholar
32 : Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry 2008; 63:642–649Crossref, Medline, Google Scholar
33 : Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA 2004; 101:10827–10832Crossref, Medline, Google Scholar
34 : Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 2007; 61:187–197Crossref, Medline, Google Scholar
35 : Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res 2005; 1037:204–208Crossref, Medline, Google Scholar
36 : Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 2002; 22:3251–3261Crossref, Medline, Google Scholar
37 : CSF and plasma pharmacokinetics of the NMDA receptor antagonist CPP after intrathecal, extradural and i.v. administration in anaesthetized pigs. Br J Anaesth 1995; 74:193–200Crossref, Medline, Google Scholar
38 : Cardiac arrest and resuscitation alters the pharmacokinetics of MK-801 in the rat. Res Commun Chem Pathol Pharmacol 1991; 73:181–195Medline, Google Scholar
39 : Ketamine. Handb Exp Pharmacol 2008; 182:313–333Crossref, Medline, Google Scholar
40 : Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol Psychiatry (Epub ahead of print, Feb 28, 2012)Google Scholar
41 : The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 2012; 13:22–37Crossref, Google Scholar
42 : Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci 2010; 30:1363–1376Crossref, Medline, Google Scholar
43 : A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci 2009; 12:751–758Crossref, Medline, Google Scholar
44 : An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 2005; 45:563–573Crossref, Medline, Google Scholar
45 : Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron 2012; 73:121–134Crossref, Medline, Google Scholar
46 : v-SNARE composition distinguishes synaptic vesicle pools. Neuron 2011; 71:474–487Crossref, Medline, Google Scholar
47 : VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci 2012; 15:738–745Crossref, Medline, Google Scholar

