
Brain-Derived Neurotrophic Factor Signaling and Subgenual Anterior Cingulate Cortex Dysfunction in Major Depressive Disorder
Abstract
Objective
The subgenual anterior cingulate cortex is implicated in the pathology and treatment response of major depressive disorder. Low levels of brain-derived neurotrophic factor (BDNF) and reduced markers for GABA function, including in the amygdala, are reported in major depression, but their contribution to subgenual anterior cingulate cortex dysfunction is not known.
Method
Using polymerase chain reaction, we first assessed the degree to which BDNF controls mRNA expression (defined as BDNF dependency) of 15 genes relating to GABA and neuropeptide functions in the cingulate cortex of mice with reduced BDNF function (BDNF-heterozygous [Bdnf+/−] mice and BDNF exon-IV knockout [BdnfKIV] mice). Gene expression was then quantified in the subgenual anterior cingulate cortex of 51 postmortem subjects with major depressive disorder and comparison subjects (total subjects, N=102; 49% were women) and compared with previous amygdala results.
Results
Based on the results in Bdnf+/− and BdnfKIV mice, genes were sorted into high, intermediate, and no BDNF dependency sets. In postmortem human subjects with major depression, BDNF receptor (TRKB) expression, but not BDNF, was reduced. Postmortem depressed subjects exhibited down-regulation in genes with high and intermediate BDNF dependency, including markers of dendritic targeting interneurons (SST, NPY, and CORT) and a GABA synthesizing enzyme (GAD2). Changes extended to BDNF-independent genes (PVALB and GAD1). Changes were greater in men (potentially because of low baseline expression in women), displayed notable differences from prior amygdala results, and were not explained by demographic or clinical factors other than sex.
Conclusions
These parallel human/mouse analyses provide direct (low TRKB) and indirect (low expression of BDNF-dependent genes) evidence in support of decreased BDNF signaling in the subgenual anterior cingulate cortex in individuals with major depressive disorder, implicate dendritic targeting GABA neurons and GABA synthesis, and, together, suggest a common BDNF-/GABA-related pathology in major depression with sex- and brain region-specific features.
Major depressive disorder is a debilitating disorder of low affect and altered mood regulation that affects approximately 17% of the population at some point in life, resulting in serious personal, social, and economic burdens (1). The prevalence of major depressive disorder is two times higher in women than in men. Female patients with the disorder tend to have higher symptom numbers, a more severe type of depression, and greater risk of recurring episodes compared with male patients, but the underlying biological vulnerabilities have not been characterized (2). Changes in the structure, function, and coordinated activity of several brain regions may underlie impaired mood regulation in depression (3). Increased metabolic activity in one of these regions, the subgenual anterior cingulate cortex, has been consistently reported in the induction of the depressive state, and subgenual anterior cingulate cortex metabolism is reversed by pharmacological treatment (4) and deep brain stimulation (5).
Low neurotrophic support in limbic brain regions has been proposed as a unifying hypothesis for the reduced density or cell numbers in the frontal cortex (6) and amygdala (7) and the reduced hippocampal volume observed in individuals with major depression (8). Rodent studies have demonstrated that various antidepressant treatments increase brain-derived neurotrophic factor (BDNF) expression (9), and BDNF infusion into the hippocampus is sufficient to produce an antidepressant-like effect (10). Despite abundant animal studies supporting the close relationship between BDNF and depression, direct evidence in humans is limited to reports of low circulating peripheral BDNF levels, which are normalized by antidepressant treatment (11), and studies demonstrating reduced pro-BDNF and BDNF levels in the postmortem amygdala of depressed female subjects (12) and in the hippocampal tissue in depressed patients (13, 14). Additionally, studies have reported that individuals who die by suicide exhibit low hippocampal and midbrain BDNF levels (15), reduced activity-dependent BDNF expression by hypermethylation of promoter/exon IV of the BDNF gene (16), and, in carriers of the BDNF Met allele, increased risk for violent suicide (17, 18), together providing additional evidence that BDNF has a role in the psychopathology of major depression.
In parallel, human imaging and basic science studies have suggested excitation/inhibition impairment in individuals with major depression that is potentially mediated by decreased GABA content (19). We recently reported down-regulation of several GABA-related genes in the dorsolateral prefrontal cortex (20), subgenual anterior cingulate cortex (21), and amygdala (12) in patients with major depression, potentially affecting somatostatin-positive dendritic targeting interneurons. We further demonstrated that a set of amygdala-related gene changes (affecting the TAC1, CORT, NPY, SST, RGS4, and SNAP25 genes [Table 1]) correlate with reduced BDNF expression in depressed patients and in mice with reduced BDNF function, hence identifying a pattern of reduced BDNF-dependent gene expression in major depression (12) and providing supporting evidence for a link between the neurotrophic (9) and GABA (19) hypotheses implicated in depression.
| Gene Code | Gene Name | Bdnf+/− Mice | BdnfKIV Mice | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Combined Male-Female | Male | Female | Combined Male-Female | Male | Female | ||||||||
| Average Log Ratiob | p | Average Log Ratiob | p | Average Log Ratiob | p | Average Log Ratiob | p | Average Log Ratiob | p | Average Log Ratiob | p | ||
| Bdnf-IX | Brain-derived growth factor | −0.56† | 0.02† | −0.74† | 0.01† | −0.34 | 0.17 | −0.70† | 0.02† | −0.62† | 0.03† | −0.80‡ | 0.001‡ |
| Trkb (Ntrk2) | Tyrosine kinase receptor | 0.03 | 0.43 | −0.01 | 0.48 | 0.07 | 0.34 | −0.08 | 0.24 | −0.02 | 0.45 | −0.16 | 0.07 |
| High BDNF dependency | |||||||||||||
| Cort | Cortistatin | −0.68‡ | 0.001‡ | −0.64‡ | 0.002‡ | −0.72† | 0.04† | −1.30‡ | 1.9E-04‡ | −0.92† | 0.05† | −1.82‡ | 4.4E-06‡ |
| Vgf | Neurotrophic growth factor inducible | −0.64‡ | 0.001‡ | −0.56† | 0.04† | −0.74‡ | 0.002‡ | −0.24 | 0.10 | −0.53† | 0.02† | 0.15 | 0.26 |
| Sst | Somatostatin | −0.59‡ | 0.003‡ | −0.41† | 0.01† | −0.80† | 0.004† | −0.60‡ | 1.4E-04‡ | −0.64‡ | 0.003‡ | −0.55† | 0.01† |
| Tac1 | Protachykinin-1 | −0.73‡ | 5.1E-05‡ | −0.66‡ | 0.002‡ | −0.81‡ | 0.001‡ | −0.82‡ | <0.001‡ | −0.70† | 0.03† | −1.00‡ | 4.7E-05‡ |
| Npy | Neuropeptide Y | −0.55† | 0.01† | −0.45† | 0.05† | −0.68† | 0.05† | −0.42‡ | 0.001‡ | −0.47† | 0.01† | −0.34† | 0.03† |
| Intermediate BDNF dependency | |||||||||||||
| Snap25 | Synaptosomal-associated protein 25 | −0.37† | 0.04† | −0.44† | 0.04† | −0.29 | 0.20 | 0.02 | 0.44 | −0.27 | 0.11 | 0.43‡ | 0.004‡ |
| Gad2 (Gad65) | Glutamate decarboxylase 2 | −0.33† | 0.04† | −0.54† | 0.02† | −0.08 | 0.32 | −0.02 | 0.46 | −0.10 | 0.33 | 0.10 | 0.12 |
| Low or no BDNF dependency | |||||||||||||
| Gad1 (Gad67) | Glutamate decarboxylase 1 | −0.09 | 0.30 | −0.33† | 0.03† | 0.19 | 0.25 | 0.03 | 0.39 | 0.16 | 0.12 | 0.29† | 0.05† |
| Pvalb | Parvalbumin | 0.03 | 0.44 | −0.35 | 0.1 | 0.47 | 0.07 | −0.09 | 0.22 | 0.19 | 0.16 | 0.04 | 0.39 |
| Rgs4 | Regulator of G protein signaling 4 | −0.13 | 0.36 | −0.33† | 0.04† | 0.11 | 0.45 | −0.05 | 0.32 | 0.04 | 0.41 | −0.18 | 0.14 |
| Slc6a1 | GABA transporter 1 | −0.31 | 0.10 | −0.42 | 0.03 | −0.17 | 0.35 | 0.10 | 0.28 | −0.10 | 0.35 | 0.39† | 0.02† |
| Calb2 | Calretinin | 0.32 | 0.32 | −0.48 | 0.10 | 1.25 | 0.15 | 0.20 | 0.18 | 0.29 | 0.20 | 0.09 | 0.32 |
| Gabra1 | GABA-A receptor, alpha 1 | 0.13 | 0.16 | 0.05 | 0.40 | 0.23 | 0.1 | 0.00 | 0.5 | 0.09 | 0.23 | −0.12 | 0.22 |
In the present study, we investigated molecular evidence for a low BDNF and reduced GABA function pathway in the subgenual anterior cingulate cortex in individuals with major depressive disorder. We first tested the degree of BDNF dependency on gene expression of a set of GABA- and BDNF-related genes in the cingulate cortex of mice with reduced BDNF function. Our choice of genes was determined by our previous study of the amygdala in depressed postmortem subjects (12) to enable comparative analyses across the two studies. Based on this information, we assessed changes in three sets of genes, with high, intermediate, or no BDNF dependency, in human subgenual anterior cingulate cortex using postmortem brain samples from a large cohort of subjects with major depressive disorder and matched comparison subjects. Exploratory analyses were performed on putative sex differences in expression patterns, given the greater female vulnerability and the previous findings of more robust somatostatin down-regulation in women with major depression (21).
Method
Human Postmortem Subjects
After consent from the next of kin, brain samples were obtained during autopsies performed at the Allegheny County Medical Examiner’s Office (Pittsburgh) using procedures approved by the University of Pittsburgh Institutional Review Board and the Committee for Oversight of Research Involving the Dead. Consensus DSM-IV diagnoses were made by an independent committee of experienced clinical research scientists using information from clinical records, toxicology results, and standardized psychological autopsies (22). Fifty-one pairs of subjects were analyzed. Each pair consisted of one subject with major depressive disorder and a comparison subject matched for sex; group means for age, postmortem interval, and brain pH were nearly identical (see Table S1 in the data supplement accompanying the online edition of this article). Subgenual anterior cingulate cortex samples containing all six cortical layers were harvested from coronal sections as described elsewhere (23).
Mice
BDNF-heterozygous (Bdnf+/−) mice (3–4 months old) were bred on a mixed S129/Sv×C57BL/6 genetic background (24). BDNF exon-IV knockout (BdnfKIV) mice were crossed on C57BL/6 as described elsewhere (25). All mice were maintained under standard conditions (i.e., in a 12/12-hour light-dark cycle, in 22°C [SD=1], and with food and water ad libitum). Brains were rapidly removed and flash frozen on dry ice. The left and right cingulate cortices were micropunctured using 0.5-mm diameter punches (23) and stored in TRIzol (Invitrogen, Carlsbad, Calif.) at –80°C. All animal care and treatment was in accordance with the Guide for Care and Use of Laboratory Animals established by the National Institutes of Health.
Real-Time Quantitative Polymerase Chain Reaction (PCR)
Total RNA was isolated from TRIzol homogenates of the subgenual anterior cingulate cortex in all 51 pairs of postmortem subjects (major depression and comparison subjects) and of the cingulate cortex in rodents. The samples were purified using RNeasy spin columns (Valencia, Calif.), and RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Walbronn, Germany). To generate cDNA, 1 μg total RNA was mixed with oligo-dT primers and SuperScript II reverse transcriptase (Invitrogen, Carlsbad, Calif.) per the manufacturer’s protocol. PCR products were amplified in quadruplets on a Mastercycler real-time PCR machine (Eppendorf, Hamburg, Germany) using universal PCR conditions as described elsewhere (21). Results were calculated as the geometric mean of threshold cycles normalized to three validated internal controls (actin, glyceraldehyde-3-phosphate dehydrogenase, and cyclophilin G).
Gene Selection
We assessed mRNA expression of several genes that we previously demonstrated to be affected in the amygdala in individuals with major depression and that display various levels of BDNF dependency, based on expression levels in the amygdala in the same strains of mice with reduced BDNF functions (Bdnf+/− and BdnfKIV) used in the present study (24, 25). Our set includes genes related to BDNF signaling (BDNF and TRKB), BDNF-dependent genes (CORT, NPY, SST, VGF, TAC1, SNAP25, and RGS4), and GABA-associated genes (GAD1, GAD2, GABRA1, SLC6A1, CALB2, PVALB, SST, NPY, and CORT) (Table 1).
Protein Isolation and BDNF Measurements
Following RNA extraction, acetone precipitation of proteins was carried out from the TRIzol samples, and Western blot analysis was performed as described elsewhere (23). Dual signals were detected using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, Neb.), and BDNF signal ratios to actin were calculated. Samples were processed in matched pairs on the same gel, and results were replicated for a total of three different Western blots. Test assays were run with 5–50 μg of total protein using the following antibodies: mouse antihuman BDNF (R&D Systems, Minneapolis), antihuman TrkB (sc-8316; Santacruz Biotechnology, Santa Cruz, Calif.), and antiactin (A2228; Sigma-Aldrich, St. Louis). We also measured BDNF using an enzyme-linked immunosorbent assay (ELISA) kit (Promega, Madison, Wisc.).
Statistical Analysis
Differences in diagnosis-dependent gene expression were determined by analysis of covariance (ANCOVA) using SPSS (SPSS, Chicago). To determine which covariates to include in the gene-specific models, each nominal factor was tested as the main factor using analysis of variance (ANOVA), scale covariates were tested using Pearson’s correlation, and repeated measures were corrected using the modified Holm-Bonferroni method (see Table S1 in the online data supplement). Samples were analyzed for the presence of antidepressant medication at the time of death. Given the small number of observations (N=9), benzodiazepines were not formally analyzed. No other psychotropic medications were present at the time of death. ANCOVA models, including significant cofactors, were then applied to the combined and sex-specific analyses. For the rodent analyses, ANOVA models with sex as a cofactor were applied to the combined analysis, and unpaired two-tailed t tests were performed in the separate male and female analyses. Values were then adjusted across genes for multiple testing using a second modified Holm-Bonferroni test (i.e., false discovery rate-corrected). Uncorrected p values for diagnosis effects are listed in Table 1.
Results
BDNF-Dependent Gene Expression in the Cingulate Cortex in Mice With Altered BDNF Function
Since BDNF expression and function vary significantly depending on brain regions, sex, and cellular activity, we first tested the role of BDNF in regulating the expression of the genes of interest in the cingulate cortex of mice that were heterozygous for a constitutive deletion of the Bdnf gene (Bdnf+/−) or that had a targeted disruption of exon IV (BdnfKIV), with the latter mutation resulting in blockade of activity-dependent BDNF protein expression (25). Bdnf mRNA levels were reduced in Bdnf+/− mice (Table 1). When the groups were separated by sex, this decrease reached statistical significance only in male mice, potentially reflecting reduced analytical power in smaller groups. Bdnf mRNA levels were also reduced in BdnfKIV mice, in both the combined and sex-specific groups (Table 1). These latter results reveal that constitutive and activity-dependent functions of BDNF are not only reduced in the cingulate cortex in BdnfKIV mice, which is consistent with an independent study of the same BdnfKIV mouse strain reporting activity-dependent BdnfKIV promoter-driven transcription abolishment in the cortex (26), but they are also significantly reduced in basal transcription by promoters I, II, III, VI, and IXa. The levels of Trkb, the main receptor of Bdnf, were not affected in the two mouse models.
Using the Bdnf+/− and BdnfKIV mouse models, we next investigated the degree of BDNF regulation on target genes (defined as BDNF dependency) within the cingulate cortex. The quantitative PCR (qPCR) analysis revealed robust and significant decreases in gene transcript levels for Cort, Vgf, Sst, Tac1, and Npy expression (false discovery rate-corrected significance in the combined male-female group in at least one strain of mice) but less robust effects on Snap25 and Gad2 expression (uncorrected significance in the combined male-female group in at least one strain of mice) and little or no effect of decreased BDNF function on Gad1, Pvalb, Rgs4, Gat1, and Gabra1 expression (no difference in the combined male-female group and uncorrected significance in the separate sex groups in one strain of mice or no change observed at all). Combined, these findings provided us with three sets of genes with a gradient of BDNF dependency in the cingulate cortex. Results of this analysis are summarized in Table 1.
BDNF-Dependent Gene Expression Changes in the Subgenual Anterior Cingulate Cortex in Postmortem Subjects With Depression
To evaluate subgenual anterior cingulate cortex BDNF function in the postmortem brain of subjects with major depression and comparison subjects, we used qPCR to measure the mRNA expression levels of BDNF, of the BDNF receptor TRKB, and of the three sets of genes with variable BDNF dependency described above. The qPCR measures were first evaluated for the effect of relevant cofactors to include in the main ANCOVA models for each gene, as described earlier. None of the investigated cofactors displayed consistent effect. The cofactor analyses and ANCOVA inclusions are summarized in Table S2 in the online data supplement.
Results from the ANCOVA analyses for the effects of major depression are summarized in Table 2. Although BDNF mRNA levels were not changed, mRNA levels of the BDNF receptor TRKB were significantly reduced by approximately 30% after correction for multiple testing. In view of the negative BDNF mRNA finding and the complex regulation of BDNF mRNA at the protein level, we attempted to measure extracts using quantitative Western blot tests and ELISA. However, the pro- and mature forms of BDNF were below detection levels in the subgenual anterior cingulate cortex samples using both approaches. Similarly, the signal-to-background ratio for TrkB protein levels was low in the samples and precluded robust quantification.
| Gene | Combined Male-Female | Male | Female | |||
|---|---|---|---|---|---|---|
| Average Log Ratioa | p | Average Log Ratioa | p | Average Log Ratioa | p | |
| BDNF | 0.04 | 0.75 | 0.01 | 0.95 | 0.08 | 0.68 |
| TRKB | −0.52‡ | 0.003‡ | −0.49† | 0.03† | −0.55† | 0.04† |
| High BDNF dependency | ||||||
| CORT | −0.58‡ | 1.69E-05‡ | −0.69‡ | 0.002‡ | −0.48‡ | 3.00E-03‡ |
| VGF | −0.63‡ | 0.001‡ | −0.81‡ | 3.76E-04‡ | −0.49 | 0.09 |
| SSTb | −0.59‡ | 0.001‡ | −0.40† | 0.04† | −0.83† | 0.009† |
| TAC1 | −0.42† | 0.02† | −0.27 | 0.22 | −0.57† | 0.02† |
| NPY | −0.66‡ | 1.01E-04‡ | −0.75‡ | 0.004‡ | −0.58‡ | 0.002‡ |
| Intermediate BDNF dependency | ||||||
| SNAP25 | −0.69‡ | 4.53E-0.4‡ | −0.98‡ | 0.001‡ | −0.39 | 0.12 |
| GAD2 (GAD65) | −0.48‡ | 2.00E-0.3‡ | −0.60‡ | 0.005‡ | −0.36 | 0.11 |
| Low or no BDNF dependency | ||||||
| GAD1 (GAD67) | −0.41‡ | 1.00E-03‡ | −0.76‡ | 4.39E-04‡ | −0.06 | 0.66 |
| PVALB (PV) | −0.58‡ | 0.005‡ | −0.76† | 0.03† | −0.41† | 0.05† |
| RGS4 | −0.42 | 0.10 | −0.22 | 0.49 | −0.63 | 0.13 |
| SLC6A1 (GAT1) | −0.06 | 0.82 | −0.11 | 0.74 | −0.01 | 0.98 |
| CALB2 (calretinin) | −0.13 | 0.23 | −0.41‡ | 0.001‡ | 0.16 | 0.38 |
| GABRA1 | 0.00 | 0.97 | −0.11 | 0.21 | 0.11 | 0.25 |
Reduced expression was identified for CORT (–33%), SNAP25 (–38%), VGF (–35%), SST (–34%), NPY (–37%), GAD1 (–25%), GAD2 (–28%), and PVALB (–33%). Reduced expression was also observed for TAC1 (–25%) but only at nominal uncorrected significance. The RGS4, GABA transporter 1/SLC6A1, calretinin/CALB2, and GABRA1 genes were unchanged.
When the groups were segregated by sex, BDNF levels remained unchanged, and TRKB was similarly reduced in both male (–29%) and female (–32%) depressed subjects compared with the respective comparison subjects, although results were less robust and only showed nominal uncorrected significance. Overall, male depressed subjects exhibited more robust decreased expression across the gene panels than female depressed subjects compared with the respective comparison subjects. Out of the eight genes displaying false discovery rate-corrected significance in the combined male-female group, six remained significant at false discovery rate-corrected p values and two at uncorrected values in the male cohort, compared with two and three genes, respectively, in the same categories in the female cohort. Out of the four genes showing no effect in the combined group, calretinin/CALB2 was decreased in the male cohort at false discovery rate-corrected significance.
When summarized by BDNF-dependent categories (Table 2), these results suggest a robust decrease in BDNF signaling in the subgenual anterior cingulate cortex in depressed subjects, with changes related to GABA functions also including genes with moderate or low BDNF dependency.
BDNF-Dependent mRNA Sex Differences in Comparison Postmortem Subjects
To investigate potential sources of the discrepancy in altered major depression-related gene expression between male and female subjects, we compared expression differences between male and female comparison subjects (i.e., individuals without psychiatric diagnoses). We found no sex differences in BDNF or TRKB mRNA expression, but several genes within the investigated panel, including CORT, NPY, CALB2, and TAC1, showed reduced baseline expression in women (Figure 1). This suggests that levels of expression in the female comparison subjects may already be closer to the low expression levels observed in depressed subjects.
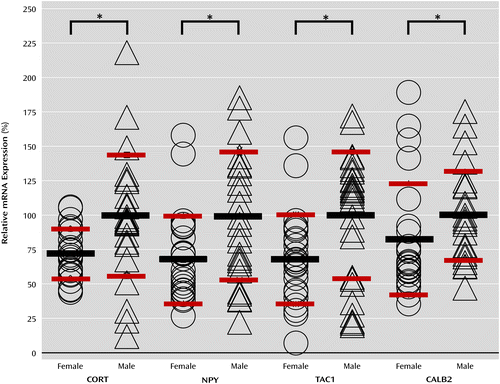
a Comparisons are between male and female comparison subjects without psychiatric diagnoses. SST mRNA expression was lower in female subjects at a level that fell short of significance (p<0.1). Black bars indicate mean values, and red bars indicate the coefficients of variation. The asterisk indicates statistical significance (p<0.05).
Discussion
Seeking molecular evidence in support of a low BDNF and reduced GABA function pathway in the subgenual anterior cingulate cortex in individuals with major depression, we first relied on two strains of mice with reduced BDNF function to determine the extent to which our genes of interest depend on BDNF for expression in the cingulate cortex (Table 1). Translating this information to human subjects with major depression and measuring expression levels in their subgenual anterior cingulate cortex, we observed no changes in BDNF itself, reduced expression of TRKB, the main receptor through which BDNF signals, and reduced mRNA levels of several genes for which expression depends on BDNF (Table 2). Among these BDNF-dependent and depression-affected genes, the reduced expression of several markers of GABA-ergic interneurons that specifically target the dendritic compartment of pyramidal neurons (SST, NPY, and CORT) suggests the presence of a reduced dendritic inhibition phenotype in individuals with major depression, downstream from low BDNF signaling. GABA-related changes also extended to genes with modest or no evidence for BDNF dependency (based on mouse studies [also see Table 1 and Table 2]), suggesting that additional factors lead to reduced GABA function in depression. Overall, results were more robust in men, which is contrary to our previous observations in the amygdala, in which depressed women exhibited reduced BDNF levels (but not TRKB levels) and greater BDNF-dependent gene changes compared with men. Together, these results suggest a core BDNF-/GABA-related pathology in major depression that affects markers of interneurons targeting pyramidal cell dendrites and that displays sex- and brain region-specific features.
With the exception of sex and, to a lesser extent, age, none of the clinical, demographic, and technical parameters had any consistent detectable effects on gene expression in our relatively large cohort of human subjects. Death by suicide has been associated with reduced BDNF expression, but differences observed in the present study appeared to be more robust in subjects who did not die by suicide, compared with the respective comparison subjects (see Table S3 in the online data supplement).
When the groups were segregated by sex, we observed similar changes in TRKB expression and overall lower statistical significance of changes in depression-related gene down-regulation in female subjects, although not systematically (Table 2). Notably, the expression levels of three BDNF-dependent genes (CORT, NPY, and TAC1) were already lower in female comparison subjects relative to male comparison subjects (Figure 1). Despite lower baseline levels, expression changes for CORT, TAC1, and NPY still displayed greater or equal statistical significance and effect size in depressed female subjects compared with depressed male subjects. Thus, despite a less robust profile of molecular changes, the low female baseline expression for some genes may result in a greater propensity to reach the threshold of low pathophysiological function (Table 2). Finally, the overall male-female similarities in gene changes downstream from low BDNF in the two mouse strains (Table 1) and the reduction or absence of changes in human female subjects for genes with intermediate and low BDNF dependency (SNAP25, GAD1/GAD2 [Table 2]) suggest the presence of additional sex-specific moderating factors in human subjects. This is consistent with our previous study of the amygdala in depressed male (23) and female (12) subjects, in which BDNF and BDNF-dependent genes were robustly affected in women but not in men. Together, these results support the concept that sex differences in the vulnerability to and the expression of major depression may not result from different pathophysiological mechanisms but rather from moderating biological factors acting on a core phenotype implicating a putative reduction in GABA function and dendritic targeting interneuron vulnerability.
SST, NPY, and CORT are three neuropeptide coding genes with overlapping patterns of expression in mice that are found in approximately 20% of interneurons and that have the functional characteristic of providing GABA-mediated inhibition to distal dendrites of pyramidal neurons. Moreover, TAC1, the fourth neuropeptide coding gene that is similarly down-regulated in the subgenual anterior cingulate cortex and amygdala in individuals with major depression (Figure 2), encodes for substance P, a gene product with putative antidepressant activity (27), which in the cortex mainly activates SST-positive cells through NK1R receptor binding (28). Since all four genes are dependent on BDNF for their expression (Table 1), low BDNF signaling may orchestrate a synergy between decreased TAC1, SST, NPY, and CORT expression, leading to reduced inhibition onto the dendritic trees of targeted pyramidal neurons. VGF and SNAP25, two genes involved in synaptic function and previously implicated in major mental illnesses, were also found to be BDNF dependent and down-regulated in the subgenual anterior cingulate cortex in subjects with major depression, suggesting that a broader BDNF-dependent module may be affected. Finally, expression of GAD1, a gene encoding an enzyme that produces GABA, and expression of PV, a gene encoding a marker for fast spiking GABA interneurons targeting the cell body and axon initial segment, were also down-regulated. For PV and GAD1, the mechanism appears to be independent of reduced BDNF function (Table 1, Table 2). Notably, PV levels were not affected in other brain regions (the dorsolateral prefrontal cortex [e.g., 20] and the amygdala [e.g., 12]), and calretinin (CALB2), a marker for a third interneuron subset, displayed reduced baseline expression and no depression-related changes in the subgenual anterior cingulate cortex but was decreased in the amygdala in depressed female subjects (Figure 2). Together, these findings put forward critical observations of the pathology of major depression, which may relate to three consecutive biological scales: 1) molecular function, manifested by altered BDNF-/TrkB- and GABA-associated gene function; 2) cellular microcircuitry, in which findings appear to be clustered by function (i.e., dendritic inhibition); and 3) circuit moderators, in which sex-related factors and brain regions are relevant modulators to gene expression in major depression. Hence, reduced GABA-mediated inhibition of incoming information in pyramidal dendrites may represent a putative microcircuitry-level phenotype underlying the increased activation of the subgenual anterior cingulate cortex and amygdala that is frequently reported in studies of patients with major depression (29, 30). In turn, restoring dendritic inhibitory function may reduce pyramidal cell activation and excitatory tone and contribute to the reduction in activation of the subgenual anterior cingulate cortex with positive treatment response to therapeutic intervention (e.g., deep brain stimulation, antidepressants) (4, 5).
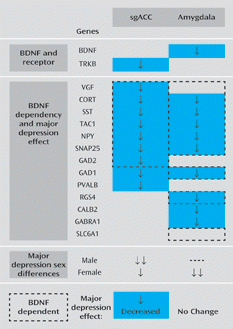
a Reduced BDNF signaling in major depressive disorder is suggested by the findings of low BDNF in the amygdala and low TrkB in the subgenual anterior cingulate cortex. Analyses of mice with reduced BDNF functions suggest that co-occurring GABA-related gene changes are partly downstream from low BDNF signaling, but the exact nature and extent of downstream gene changes are moderated by brain region- and sex-specific factors. Comparison findings in the amygdala are taken from a previous study (12). SgACC=Subgenual anterior cingulate cortex.
Some of the limitations of these results are inherent to investigation of heterogeneous cohorts and of postmortem brain samples. Large numbers of clinical, demographic, and technical parameters have to be taken into consideration, and results are mostly correlative and cannot provide insight into developmental processes in major depression. Our relatively large cohort size allowed us to rule out major effects of putative confounds (details are summarized in Table S2 in the online data supplement), but the results will need to be confirmed in independent cohorts.
The causal link between reduced BDNF signaling and altered gene expression was inferred from analyses of rodents with genetically induced reduction in BDNF function, but species differences may exist, and thus the different labels of gene-specific BDNF dependency may vary. The fact that regional variations in BDNF dependency were also observed in rodents suggests that aspects of the human gene regulation patterns are conserved across species. This latter observation supports the need for further studies of rodent models with more refined genetic manipulations affecting specific interneuron populations, for instance, to assess the effect on reduced dendritic inhibition on the local microcircuitry in the amygdala and cingulate cortex and downstream behavioral phenotypes. Indeed, it is evident that the complexity of the putative BDNF-mediated cellular and signaling phenotype observed in human major depression is not fully replicated in currently available genetic rodent models, and thus caution should be applied when interpreting rodent behavioral outputs downstream from broad genetic changes; for instance, Bdnf+/− and BdnfKIV mice exhibit normal and increased emotionality, respectively (31, 32). Disruption of forebrain-specific BDNF leads to higher emotionality when combined with exposure to chronic stress in female mice (33, 34). Conversely, low ventral striatum BDNF can have antidepressant-like effects (35). Our observation in humans of reduced expression of BDNF in the amygdala and of reduced TRKB in the subgenual anterior cingulate cortex suggests that altered BDNF signaling may represent a complex integration of currently unidentified upstream events (e.g., stress factors, developmental trajectories, and genetic variation), which result in similar core downstream changes (i.e., reduced markers of dendritic inhibition) and that are further moderated by numerous factors (e.g., sex, brain region, and brain activity).
Finally, it is also becoming evident that the observed findings are not specific to major depression, since similar reductions in BDNF and SST expression have been reported in studies of other neuropsychiatric (e.g., schizophrenia [36, 37] and bipolar disorder [38]) and neurological (e.g., Alzheimer’s disease and Huntington’s disease) disorders. Thus, our findings may more accurately reflect a molecular and cellular endophenotype that implicates BDNF signaling and GABA microcircuitry and that has its own etiological factors. However, the restricted scope on markers affecting dendritic inhibition that we observed differs from observations in studies of other diseases in which changes occur in the context of other core pathologies, such as robust PV-related GABA dysfunction in schizophrenia (39) or neurodegenerative processes in Alzheimer’s disease (40). Investigating the etiological factors and phenotypic outputs of these respective molecular and cellular endophenotypes outside the restriction of the categorical definitions of psychiatric and neurological illnesses may provide dimensional insight into relevant proximal pathophysiological mechanisms to be targeted for therapeutic purposes, while their patterns of co-occurrences may be informative of mechanisms underlying clusters of symptoms that are enriched in clinically defined disorders.
1 : The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105Crossref, Medline, Google Scholar
2 : Gender-mediated clinical features of depressive illness. The importance of temperamental differences. Br J Psychiatry 1990; 157:835–841Crossref, Medline, Google Scholar
3 : Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage 2004; 22:409–418Crossref, Medline, Google Scholar
4 : Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry 2000; 48:830–843Crossref, Medline, Google Scholar
5 : Deep brain stimulation for treatment-resistant depression. Neuron 2005; 45:651–660Crossref, Medline, Google Scholar
6 : Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry 2001; 49:741–752Crossref, Medline, Google Scholar
7 : Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 2002; 52:404–412Crossref, Medline, Google Scholar
8 : Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 2004; 161:1957–1966Link, Google Scholar
9 : The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol 2007; 18:391–418Crossref, Medline, Google Scholar
10 : Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 2002; 22:3251–3261Crossref, Medline, Google Scholar
11 : Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol Psychiatry 2011; 16:1088–1095Crossref, Medline, Google Scholar
12 : Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry 2011 (Epub ahead of print, September 13, 2011)Medline, Google Scholar
13 : Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci 2011; 36:195–203Crossref, Medline, Google Scholar
14 : Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J Psychiatr Res 2009; 43:1175–1184Crossref, Medline, Google Scholar
15 : Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 2003; 60:804–815Crossref, Medline, Google Scholar
16 : Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry 2010; 67:258–267Crossref, Medline, Google Scholar
17 : Interaction between BDNF Val66Met and childhood trauma on adult’s violent suicide attempt. Genes Brain Behav 2008; 7:314–322Crossref, Medline, Google Scholar
18 : The association between brain-derived neurotrophic factor polymorphism (BDNF Val66Met) and suicide. J Affect Disord 2011; 128:287–290Crossref, Medline, Google Scholar
19 : The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry 2011; 16:383–406Crossref, Medline, Google Scholar
20 : GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol 2011; 14:721–734Crossref, Medline, Google Scholar
21 : Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis 2011; 42:116–124Crossref, Medline, Google Scholar
22 : Normal cellular levels of synaptophysin mRNA expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 2000; 48:389–397Crossref, Medline, Google Scholar
23 : A molecular signature of depression in the amygdala. Am J Psychiatry 2009; 166:1011–1024Link, Google Scholar
24 : Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 1995; 92:8856–8860Crossref, Medline, Google Scholar
25 : Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci USA 2009; 106:5942–5947Crossref, Medline, Google Scholar
26 : Activity-dependent brain-derived neurotrophic factor expression regulates cortistatin-interneurons and sleep behavior. Mol Brain 2011; 4:11Crossref, Medline, Google Scholar
27 : Results from 2 randomized, double-blind, placebo-controlled studies of the novel NK1 receptor antagonist casopitant in patients with major depressive disorder. J Clin Psychopharmacol 2011; 31:727–733Crossref, Medline, Google Scholar
28 : Substance P and nitric oxide signaling in cerebral cortex: anatomical evidence for reciprocal signaling between two classes of interneurons. J Comp Neurol 2001; 441:288–301Crossref, Medline, Google Scholar
29 : Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 1999; 156:675–682Abstract, Google Scholar
30 : Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: related and independent features. Biol Psychiatry 2007; 61:198–209Crossref, Medline, Google Scholar
31 : Deficit in BDNF does not increase vulnerability to stress but dampens antidepressant-like effects in the unpredictable chronic mild stress. Behav Brain Res 2009; 202:245–251Crossref, Medline, Google Scholar
32 : Lack of promoter IV-driven BDNF transcription results in depression-like behavior. Genes Brain Behav 2010; 9:712–721Crossref, Medline, Google Scholar
33 : Gender-specific impact of brain-derived neurotrophic factor signaling on stress-induced depression-like behavior. Biol Psychiatry 2009; 66:84–90Crossref, Medline, Google Scholar
34 : Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 2007; 61:187–197Crossref, Medline, Google Scholar
35 : Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006; 311:864–868Crossref, Medline, Google Scholar
36 : Molecular determinants of dysregulated GABAergic gene expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 2009; 65:1006–1014Crossref, Medline, Google Scholar
37 : Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci 2005; 25:372–383Crossref, Medline, Google Scholar
38 : Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res 2011; 45:995–1004Crossref, Medline, Google Scholar
39 : Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry 2008; 165:479–489Link, Google Scholar
40 : Molecular insights into the pathogenesis of Alzheimer’s disease and its relationship to normal aging. PLoS ONE 2011; 6:e29610Crossref, Medline, Google Scholar

