
Evidence That Altered Amygdala Activity in Schizophrenia Is Related to Clinical State and Not Genetic Risk
Abstract
Objective: Although amygdala dysfunction is reported in schizophrenia, it is unknown whether this deficit represents a heritable phenotype that is related to risk for schizophrenia or whether it is related to disease state. The purpose of the present study was to examine amygdala response to threatening faces among healthy siblings of schizophrenia patients in whom a subtler heritable deficit might be observed. Method: Participants were 34 schizophrenia patients, 29 unaffected siblings, and 20 healthy comparison subjects. Blood-oxygen-level-dependent (BOLD) functional magnetic resonance imaging (fMRI) was conducted during an implicit facial information processing task. The N-back working memory task, which has been shown to elicit prefrontal cortex abnormalities in unaffected siblings of schizophrenia patients, was employed as a positive experimental control. Results: Schizophrenia patients demonstrated a deficit in amygdala reactivity to negative face stimuli and an alteration, correlated with neuroleptic drug dosage, in the functional coupling between the amygdala and subgenual cingulate. In contrast, unaffected siblings showed a pattern that was not statistically different from that of healthy comparison subjects. During the N-back working memory task, both schizophrenia patients and their unaffected siblings demonstrated a pattern of inefficient prefrontal cortex engagement, which is consistent with earlier evidence that this pattern is related to genetic risk for schizophrenia. Conclusions: These data suggest that the pathophysiological mechanism underlying the inability of individuals with schizophrenia to normally engage the amygdala in processing fearful and angry facial representations is more likely a phenomenon related to the disease state, specifically to treatment.
Face processing, which is integral to the processing of salient environmental cues during social interactions, is critically dependent on amygdala functioning (1) . Reduced amygdala response to fearful faces has been found in individuals with schizophrenia (2 , 3) . The response to fearful faces is widely used as a paradigm to examine the reactivity of the amygdala to salient stimuli, and the underlying circuit has been shown to be modulated by genes linked to temperament and emotional response—for example, the serotonin transporter SLC6A4 genotype (4 – 7) , catechol- O -methyltransferase (COMT) genotype (8 , 9) , and monamine oxidase A (MAO-A) genotype (10) . These observations raise questions regarding the nature of face processing deficits associated with schizophrenia and the extent to which these deficits are based on genetic and nongenetic factors. One method with the potential to differentiate genetic versus secondary risk factors associated with schizophrenia is to study face processing among the relatives of schizophrenia patients. Studies that have employed this method were conducted only at the behavioral level, and most of these studies focused specifically on the ability of high-risk subjects to recognize different emotional facial expressions. Although some of the studies that used small sample sizes showed no deficits among the relatives of schizophrenia patients (11 , 12) , one recent study with a large sample size reported that emotional deficits are related to genetic risk for schizophrenia (13) . Further complicating the comprehension of these deficits in schizophrenia patients is the evidence that symptoms influence the deficit in emotion recognition (14) , suggesting that this impairment may be state related. To explore this issue further, we analyzed amygdala function in a healthy high-risk group of subjects (unaffected siblings of schizophrenia patients) using an alternative strategy. We used blood-oxygen-level-dependent (BOLD) functional magnetic resonance imaging (fMRI) in combination with a task based on the implicit perceptual processing of threatening faces. Since previous studies have already established links of cognitive dysfunction to genetic susceptibility for schizophrenia (15 – 17) and the cognitive demand during perceptual processing of threatening faces may confound the understanding of the inheritance of face processing deficits (18) , the task we utilized involved minimal cognitive demands.
We hypothesized that if implicit face processing deficits associated with schizophrenia map to amygdala dysfunction as a heritable trait, these deficits should also be observed—at least to a subtler degree—in the unaffected siblings of schizophrenia patients, who are genetically at increased risk for schizophrenia. This method has successfully identified several brain-based intermediate phenotypes (19 , 20) . On the other hand, if these face processing deficits are related to a secondary effect of the disease (e.g., treatment, symptoms), they should be limited to schizophrenia patients and not observed in the unaffected siblings. In addition to measuring amygdala reactivity to threatening faces, we also measured the functional coupling of the amygdala with the anterior cingulate cortex, which is a key regulatory region for modulating affect and amygdala responsivity (7) . As a positive control experiment, we examined the heritability of BOLD fMRI response to working memory in the same subjects. This paradigm has previously revealed impairment in working memory-related function among the unaffected siblings of schizophrenia patients (21) .
Method
Participants
Schizophrenia patients and their unaffected siblings were recruited as part of the Clinical Brain Disorders Branch Sibling Study (National Institutes of Health [NIH], protocol 95-M-6150), which involved the study of neurobiological aspects of illness related to genetics. Healthy comparison subjects were recruited from the NIH Clinical Research Volunteer Program. The study was approved by the National Institute of Mental Health (NIMH) Intramural Program Institutional Review Board. All participants were assessed using the Structured Clinical Interview for DSM-IV (SCID). Exclusion criteria have been previously reported (22) . Schizophrenia patients were on a stable regimen of antipsychotic medication (atypical and conventional neuroleptics), measured as chlorpromazine equivalents, at the time of the study (23) . Four patients were not receiving any treatment at the time of the study. Only individuals of European ancestry were included in this data set in order to minimize population stratification artifacts. Siblings and healthy comparison subjects who were receiving any psychotropic pharmacological treatment were excluded. Schizophrenia patients who were receiving antidepressants were also excluded, since antidepressants have been shown to influence amygdala response (24) . For patients, the rating of symptom severity was recorded, using the Positive and Negative Syndrome Scale (PANSS) (25) , on the same day of the scan. After complete description of the study was given, written informed consent was obtained.
Experimental Paradigm
The face matching task is a simple perceptual task and has previously been shown to robustly engage the amygdala (4 , 5 , 7 , 9 , 10) . The block fMRI paradigm consists of two experimental conditions: an emotional identity face matching condition and a sensorimotor control task. The face matching task consists of five 30-second duration blocks. Blocks 1, 3, and 5 are sensorimotor blocks, and blocks 2 and 4 are emotion blocks. Each sensorimotor and emotion block consists of six 5-second duration trials. Each trial involves the presentation of two images in the lower panel and one image in the upper panel. In the six trials of each sensorimotor block, the two lower images are shapes, and the upper panel image is identical to one of the shapes in the lower panel. Subjects respond using button presses (left or right) to indicate which image in the lower panel matches the upper panel image. In the six trials of each emotion block, the lower panel consists of two faces, one angry and one afraid, derived from a standard set of pictures of facial affect (26) . The upper panel consists of one of the two faces shown in the lower panel. Subjects respond using button presses (left or right) to indicate which lower panel face matches the face in the upper panel.
Participants also performed a N-back working memory task administered using a block design, with the 2-back working memory condition alternating with a no-back control condition as described elsewhere (21) .
Data Acquisition
BOLD fMRI was performed with a General Electric Signa 3T scanner (Milwaukee, Wisc.) using parameters as previously described (gradient-echo echo-planar imaging: axial slices=24, thickness=4 mm, gap=1 mm; TR=2,000 msec, echo time=28 msec, field of view=24 cm, matrix=64×64 pixels [ 4 , 5 , 7 , 9 , 10 ]). The first four scans were discarded to allow for signal saturation.
Demographic and Behavioral Data
Analysis of variance (ANOVA) for continuous variables and chi square analysis for nonparametric variables were employed when appropriate (STATISTICA software, Statsoft, Tulsa, Okla.). In case there was no normal distribution, a nonparametric test (Kolmogorov-Smirnov Test, STATISTICA software, Statsoft, Tulsa, Okla.) was also applied.
fMRI
Images were processed using Statistical Parametric Mapping-2 (SPM2) (http://www.fil.ion.ucl.ac.uk/spm). For each subject, images were 1) realigned to the first volume in the time series to correct for head motion, 2) spatially normalized into a standard stereotactic space (Montreal Neurological Institute [MNI] template) using an affine and nonlinear (4×5×4 basis functions) transformation, 3) smoothed with an 8-mm full-width at half maximum Gaussian filter, and 4) ratio normalized to the whole brain global mean. All fMRI data were individually examined for motion artifacts, and subjects with excessive interscan motion (>2 mm translation, >1.5° rotation) were excluded.
The preprocessed data sets were then analyzed using a conservative second-level random effects model. To perform this second-level analysis, predetermined condition effects at each voxel for each subject were calculated using a t statistic, producing a contrast image of the emotional task relative to the sensorimotor control task for each subject. These individual contrast images were then used to identify main effects of the specific task for each group (schizophrenia patients, unaffected siblings, and healthy comparison subjects) using one-tailed t tests.
ANOVA, with the contrast images from each group, was performed to compare BOLD response differences in the amygdala. To find an “intermediate phenotype” effect (i.e., a nonclinical phenotype related to genetic risk for schizophrenia), the following contrasts were used: 1) schizophrenia patients + unaffected siblings > healthy comparison subjects (contrast: 0.5, 0.5, –1) and 2) schizophrenia patients + unaffected siblings < healthy comparison subjects (contrast: –0.5, –0.5, 1). To explore a “state disease-related” effect (an effect related to the disease but not genetically derived), the following contrasts were used: 1) schizophrenia patients > unaffected siblings + healthy comparison subjects (contrast: 1, –0.5, –0.5) and 2) schizophrenia patients < unaffected siblings + healthy comparison subjects (contrast: –1, 0.5, 0.5). We also performed three separate t tests as post hoc analyses between schizophrenia patients and healthy comparison subjects, schizophrenia patients and unaffected siblings, and unaffected siblings and healthy comparison subjects.
In addition, we performed a simple regression analysis using SPM2 entering the single-subject contrasts, with diagnosis as a predictor (schizophrenia patients=1; unaffected siblings=2; healthy comparison subjects=3). ANOVA, with gender and diagnosis as cofactors of interest, was also performed using the individual contrast images (six groups: male and female subjects in each diagnostic group).
Functional Connectivity Analysis
We measured functional connectivity employing methods previously described (7 , 10) . Utilizing a bilateral amygdala-specified mask as the region (volume) of interest, we examined the covariation of activation across the brain with activation in the amygdala during the face matching task. After mean and drift correction of the time series, median activity within the region of interest was calculated for each scan during the trials of interest (match blocks) and then correlated across the brain with all voxels, resulting in a map that contained in each voxel the correlation coefficient of the time series for a particular voxel with that of the reference region (bilateral amygdala). To avoid confounding the connectivity measures by coactivation, calculations were performed after estimated effects of the block design task were removed. The r values were then transformed into z scores using a Fisher’s r to z transform (27) . These z maps (one per subject) were analyzed using a conservative second-level random effects model (via SPM2) identical to the model we described previously. Thus, functional connectivity between the amygdala and the subgenual (Brodmann’s area 25) and supragenual (Brodmann’s area 32) anterior cingulate cortices was compared among the three groups (schizophrenia patients, unaffected siblings, and healthy comparison subjects). In addition, we performed a simple linear regression analysis entering the single-subject contrasts, with diagnosis as a predictor.
Moreover, to control for the confounding effects of other sources of variance, such as cardiovascular and respiratory noises on the connectivity measures, calculations were performed after covarying out the estimated effects of the following variables: 1) the mean CSF signal computed from the lateral ventricles (anatomical region of interest determined using Wake Forest University PickAtlas Toolbox, Version 2.0 [Winston-Salem, N.C.; http://www.fmri.wfubmc.edu]); and 2) the mean deep white matter signal estimated from two regions of interest (two 5-mm radius spheres located in the anterior corona radiata and centered on the MNI coordinates [x,y,z] 26, 23, 18 and –26, 23, 18, respectively).
Statistical Inference
For the main effect of task in each group, a threshold of p<0.05, corrected for multiple comparisons (false discovery rate; cluster extent in voxels=k>5) across the whole brain, was employed. For all other imaging data, the significance threshold was set at p<0.05, corrected for multiple comparisons (false discovery rate=k>5) within the region of interest (faces task: amygdala; Brodmann’s areas 25, 32; N-back task: Brodmann’s areas 9, 46) as defined using Wake Forest University PickAtlas Toolbox, Version 2.0.
Correlations With Variables Related to the State of Disease
To examine the effect of “state” variables (e.g., antipsychotic dose, PANSS scores) on the variability in amygdala reactivity and amygdala-cingulate coupling for schizophrenia patients with available clinical data, we performed simple correlation analysis using SPM2, with chlorpromazine equivalents or PANSS subscale scores as covariates of interest. Through the use of STATISTICA software, we conducted a correlation analysis outside of the image space between these “state” variables and the BOLD signal extracted from voxels in the amygdala and cingulate region of interest with the highest z scores.
Genotyping
To control for the possibility that the three groups might differ on allele frequencies of specific polymorphisms of the SLC6A4 , COMT, and MAO-A genotypes, which have previously been shown to affect amygdala responsivity, genotypes were determined as described previously (4 , 10 , 22) . DNA isolation and analysis were conducted on blood samples obtained from all subjects who gave informed consent according to NIMH Institutional Review Board guidelines.
Results
Demographic and Behavioral Data
All three groups (schizophrenia patients, unaffected siblings, healthy comparison subjects) were well matched for age, gender, and handedness ( Table 1 ). Schizophrenia patients differed from unaffected siblings and healthy comparison subjects on IQ and education. The three groups did not differ significantly on the distribution of the SLC6A4 genotype, MAO-A variable number of tandem repeat genotype, or COMT valine-to-methionine (Val 158 Met) genotype (all p values >0.8). For both the control and faces conditions, all subjects had a >90% average of correct responses ( Table 1 ), with no significant differences between groups (all p values >0.10).
BOLD fMRI
Main effect of the task
As previously reported (4 , 5 , 7 , 9 , 10) , the main effect of task showed a significant BOLD response in the fear network, including the amygdala-hippocampus complex, the posterior fusiform gyrus, and the prefrontal cortex bilaterally (p<0.05 false discovery rate-corrected for whole brain; k>5 for all three groups [data not included]).
Between-group analyses
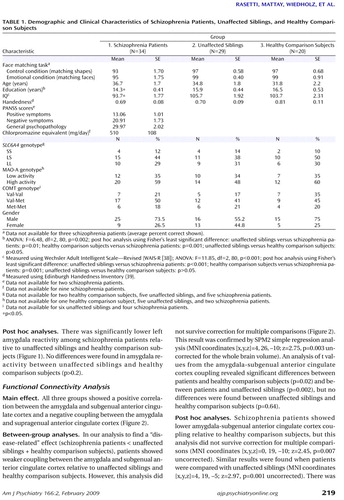
ANOVA showed a significant main effect of diagnosis on amygdala activation (F=10.13, df=2, 80, p=0.005 false discovery rate-corrected; MNI coordinates [x,y,z]=–30, 0, –25). There was a significant “disease-related” effect, with lower amygdala reactivity among schizophrenia patients relative to unaffected siblings and healthy comparison subjects ( Figure 1 ). Amygdala activity in unaffected siblings did not differ from a normal distribution (Shapiro-Wilk W test: W=0.96, p=0.27). The contrast testing for heritability (i.e., to look for an “intermediate phenotype” effect [schizophrenia patients + unaffected siblings < healthy comparison subjects]) did not show a significant effect on amygdala reactivity. All of these results were further corroborated in the SPM2 simple regression analysis using diagnosis as a covariate of interest (MNI coordinates [x,y,z]=–30, 0, –25; z=3.83, p=0.002 false discovery rate-corrected within amygdala region of interest; post hoc analysis for extracted values: schizophrenia patients versus healthy comparison subjects: p<0.001; schizophrenia patients versus unaffected siblings: p<0.001; unaffected siblings versus healthy comparison subjects: p=0.76).
a The image in the top row illustrates the “disease-related” effect (contrast: patients < unaffected siblings + healthy comparison subjects) in the left amygdala (MNI coordinates [x,y,z] –30, 0, –25; ANOVA: z=4.23, p<0.001 false discovery rate-corrected within region of interest, superimposed on a coronal slice of the T1 MNI single subject). The scatterplot in the top row illustrates amygdala activation in schizophrenia patients, unaffected siblings, and healthy comparison subjects. Post hoc analysis revealed the following significant differences: schizophrenia patients versus unaffected siblings: p=0.001; schizophrenia patients versus healthy comparison subjects: p=0.003. Amygdala activation was extracted as the first eigenvalue of the weighted parameter estimates. The bottom row illustrates post hoc analyses of the left amygdala (left contrast: schizophrenia patients <healthy comparison subjects; MNI coordinates [x,y,z] –30, 0, –25; two-sample t test: z=3.44, p=0.007 false discovery rate-corrected within the region of interest; right contrast: schizophrenia patients <unaffected siblings; MNI coordinates [x,y,z,] –30, 0, –25; two-sample t test: z=3.49, p=0.006 false discovery rate-corrected within the region of interest; L=left).
Post hoc analyses
There was significantly lower left amygdala reactivity among schizophrenia patients relative to unaffected siblings and healthy comparison subjects ( Figure 1 ). No differences were found in amygdala reactivity between unaffected siblings and healthy comparison subjects (p>0.2).
Functional Connectivity Analysis
Main effect
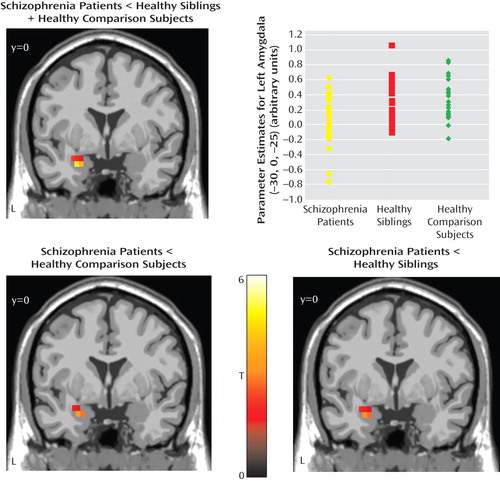
All three groups showed a positive correlation between the amygdala and subgenual anterior cingulate cortex and a negative coupling between the amygdala and supragenual anterior cingulate cortex ( Figure 2 ).
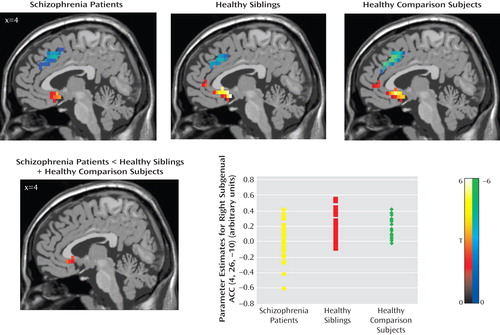
a The images in the top row illustrate the main effect of functional coupling. All p values are false discovery rate-corrected within the region of interest (Brodmann’s areas 25 and 32) superimposed on a sagittal slice of the T1-MNI single subject. Positive (red-yellow) and negative (blue-green) coupling between the bilateral amygdala and subgenual (Brodmann’s area 25) and supragenual (Brodmann’s area 32) regions, respectively, are shown for all three groups: 1) schizophrenia patients (left subgenual: MNI coordinates [x,y,z]: –4, 11, –10; z=3.66, p=0.004; right subgenual: MNI coordinates [x,y,z]: 4, 11, –10; z=3.37, p=0.007; right supragenual: MNI coordinates [x,y,z]: 4, 15, 40; z=3.80, p=0.02); 2) unaffected siblings (left subgenual: MNI coordinates [x,y,z]: –4, 8, –15; z=5.29, p<0.0001; right subgenual: MNI coordinates [x,y,z]: 4, 8, –15; z=4.72, p<0.0001; right supragenual: MNI coordinates [x,y,z]: 4, 23, 35; z=3.50, p=<0.0001); and 3) healthy comparison subjects (left subgenual: MNI coordinates [x,y,z]: –4, 11, –10; z=4.57, p<0.0001; right subgenual: MNI coordinates [x,y,z]: 4, 19, –10; z=4.13, p=0.001; left supragenual: MNI coordinates [x,y,z]: –4, 26, 35; z=4.93, p<0.0001; right supragenual: MNI coordinates [x,y,z]: 4, 26, 35; z=4.93, p<0.0001). The image in the bottom row illustrates ANOVA for “disease-related” effect (contrast: schizophrenia patients <unaffected siblings + healthy comparison subjects; right subgenual anterior cingulate cortex: MNI coordinates [x,y,z]: 4, 26, –10; z=3.01, p=0.001 uncorrected for the whole brain). The scatterplot in the bottom row illustrates amygdala-subgenual coupling in the three groups. Post hoc analysis revealed the following significant differences: schizophrenia patients versus unaffected siblings: p=0.005; schizophrenia patients versus healthy comparison subjects: p=0.031. Amygdala-subgenual coupling was extracted as the first eigenvalue of the weighted parameter estimates.
Between-group analyses
In our analysis to find a “disease-related” effect (schizophrenia patients < unaffected siblings + healthy comparison subjects), patients showed weaker coupling between the amygdala and subgenual anterior cingulate cortex relative to unaffected siblings and healthy comparison subjects. However, this analysis did not survive correction for multiple comparisons ( Figure 2 ). This result was confirmed by SPM2 simple regression analysis (MNI coordinates [x,y,z]=4, 26, –10; z=2.75, p=0.003 uncorrected for the whole brain volume). An analysis of t values from the amygdala-subgenual anterior cingulate cortex coupling revealed significant differences between patients and healthy comparison subjects (p=0.02) and between patients and unaffected siblings (p=0.002), but no differences were found between unaffected siblings and healthy comparison subjects (p=0.64).
Post hoc analyses
Schizophrenia patients showed lower amygdala-subgenual anterior cingulate cortex coupling relative to healthy comparison subjects, but this analysis did not survive correction for multiple comparisons (MNI coordinates [x,y,z]=0, 19, –10; z=2.45, p=0.007 uncorrected). Similar results were found when patients were compared with unaffected siblings (MNI coordinates [x,y,z]=4, 19, –5; z=2.97, p=0.001 uncorrected). There was no significant difference between unaffected siblings and healthy comparison subjects in amygdala-subgenual anterior cingulate cortex coupling. In our analysis to determine an “intermediate phenotype” effect, no differences in the subgenual area were found. Interestingly, in the patient group, chlorpromazine equivalent doses and amygdala-subgenual anterior cingulate cortex coupling were negatively correlated, using SPM2 simple regression analysis (MNI coordinates [x,y,z]=4, 11, –15; z=3.35, p=0.01 false discovery rate-corrected within region of interest [correlation analysis outside image space is shown in Figure 3 ]). Similar results were yielded when time courses extracted from white matter and CSF voxels were used as nuisance regressors (see the data supplement accompanying the online version of this article).
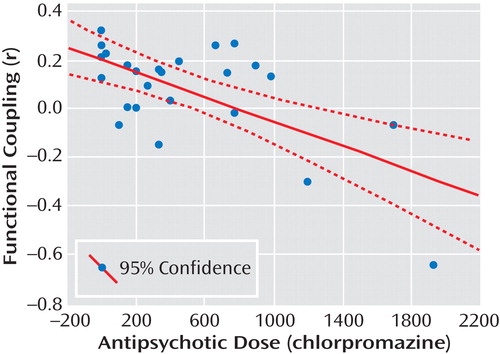
a The scatterplot illustrates findings in the right subgenual anterior cingulate cortex of functional coupling with the amygdala, which negatively correlated with antipsychotic dose (r=–0.66, p<0.001).
Analysis in a Subsample of Male Subjects
Although gender distribution was not significantly different across the three groups, nearly 50% of the subjects in the unaffected sibling group were women. However, women represented less than one-third of subjects in the patient and healthy comparison groups. To ensure that the results were not an artifact of this gender distribution, we performed additional analysis for a sample comprised only of the male subjects in each group (N=56). This analysis also revealed results similar to that of the whole group analysis (see the data supplement accompanying the online version of this article). Moreover, an ANOVA of the entire sample, using gender as a covariate, revealed no significant gender effect or gender-by-diagnosis interaction.
N-Back Working Memory Task
Schizophrenia patients performed significantly worse than their unaffected siblings and healthy comparison subjects (all p values <0.001 [ Figure 4 ]). Although unaffected siblings did not perform significantly less accurate, they showed a tendency toward significance for slower reaction time relative to healthy comparison subjects (p=0.1), but no difference for reaction time was observed between unaffected siblings and schizophrenia patients (p=0.86). To control for accuracy differences, fMRI was conducted for subjects with performance-matched group data (N=53), which revealed that patients and unaffected siblings had inefficient dorsolateral prefrontal cortex response (i.e., greater activation for a fixed level of performance) relative to healthy comparison subjects (see Figure 4 and the data supplement accompanying the online version of this article). Analysis of covariance for unaffected siblings and healthy comparison subjects, using reaction time as a covariate of no interest, once again revealed a significant difference, with greater dorsolateral prefrontal cortex activation among unaffected siblings relative to healthy comparison subjects (MNI coordinates [x,y,z]=–30, 22, 38; z=3.66, p=0.04 false discovery rate-corrected within region of interest; Brodmann’s areas 9, 46).
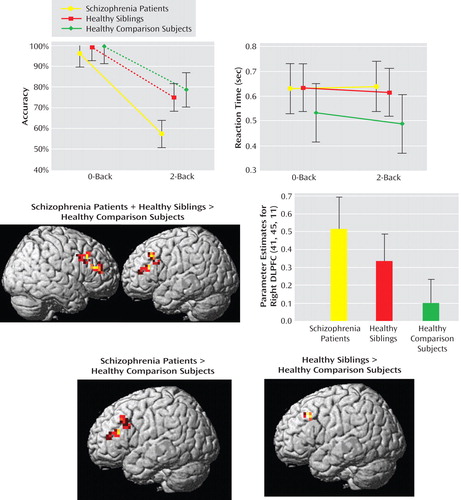
a The top row illustrates performance data for the entire sample (data available for 60 subjects). The images in the middle row illustrate ANOVA for the performance-matched sample (data available for 53 subjects). (“intermediate phenotype” effect; contrast: schizophrenia patients + unaffected siblings > healthy comparison subjects). The graph in the middle row illustrates mean ± 95% confidence intervals. Parameter estimates were extracted from the right dorsolateral prefrontal cortex. Post hoc analysis showed the following significant differences: patients versus healthy comparison subjects: p=0.0008; siblings versus healthy comparison subjects: p=0.03; patients versus siblings: p=0.09. The images in the bottom row illustrate post hoc analysis. The maps were thresholded at a significance level of p<0.05, uncorrected within the region of interest Brodmann’s areas 9, 46 only for illustrative purpose.
A two-sample t test conducted for the group of subjects for whom N-back data were available and the group of subjects for whom N-back data were not available revealed no differences in amygdala reactivity during the face matching task between the groups.
Discussion
In the present study, schizophrenia patients demonstrated decreased amygdala reactivity in response to potentially threatening stimuli as well as a decrease in amygdala-subgenual cingulate functional coupling relative to their unaffected siblings and healthy comparison subjects. Schizophrenia patients also showed a negative correlation between antipsychotic dose and the amygdala-subgenual cingulate functional coupling. However, no differences in the amygdala response between unaffected siblings and healthy comparison subjects were found.
Our finding of decreased amygdala reactivity suggests that schizophrenia patients have a deficit in the recruitment and regulation of limbic areas implicated in the response to salient stimuli, specifically fear. This finding is consistent with several previous reports (3) . Canli et al. (28 , 29) reported that left amygdala reactivity is linked to the subjective ratings of the emotional intensity of pictures. Phelps et al. (30) suggested that the left amygdala modulates fear response to a mental representation of an aversive event. Based on these previous findings, the left amygdala hypoactivation observed in our patient sample may reflect reduced threat sensitivity or a deficit in more abstract representations of threat.
Recent studies of healthy volunteers (7 , 10 , 31) have shown a pattern of activation/deactivation of limbic and paralimbic regions during face processing, implicating a hypothetical model for regulation of amygdala reactivity. According to this model (7) , reduced coupling is the causal proximal event in this regulatory circuit, and reduced amygdala-cingulate connectivity thereby leads to increased amygdala reactivity. In the present study, all three groups (schizophrenia patients, unaffected siblings, and healthy comparison subjects) showed this proposed pattern of activation/deactivation of limbic and paralimbic regions. However, in contrast to the prediction that decreased coupling would be associated with increased amygdala reactivity, we observed a tendency toward weaker coupling between the amygdala and the subgenual anterior cingulate cortex in schizophrenia patients, despite reduced amygdala reactivity. This raises the possibility that the decreased amygdala activity in patients results from a primary dysfunction within the amygdala rather than from abnormal cortical regulation. Our data also suggest that there is an effect of antipsychotic treatment on this circuit, suggesting that the deficit may be state related.
In contrast to our findings for schizophrenia patients, we failed to detect any differences in amygdala reactivity and amygdala functional coupling with other brain regions between unaffected siblings and healthy comparison subjects. The possibility that our null finding—between unaffected siblings and healthy comparison subjects during the processing of threatening faces—was the result of something atypical about our sample of siblings appears to be unlikely, since the expected abnormal prefrontal response to an executive cognition task was observed in a subsample of these same siblings. Since we demonstrated an influence of treatment on amygdala coupling, our null finding in the amygdala between unaffected siblings and healthy comparison subjects suggests that the deficits in amygdala reactivity observed in schizophrenia patients are unlikely related to genetic risk for schizophrenia but more likely related to state factors.
To our knowledge, only one study, conducted by Habel et al. (32) , has examined the emotional circuit in the unaffected siblings of schizophrenia patients using functional neuroimaging. Habel et al. employed a mood induction task and reported a reduction in amygdala reactivity for both schizophrenia patients and their unaffected siblings relative to healthy comparison subjects. However, the facial emotion processing task of the Habel et al. study utilized sad and happy faces. Thus, differences in the emotional valences and cognitive demand employed in the present study versus the study conducted by Habel et al. may be responsible for the different results.
Our finding of decreased amygdala activity in schizophrenia patients is in accordance with several previous reports in the literature (3) . However, there are a few studies (33 – 35) that have reported increased activity in medial temporal lobe structures, including the amygdala. These conflicting results could possibly be explained by a number of factors, including drug status, antipsychotic dose, genetic variability of patients in the different studies, and different task designs.
There are some limitations to our study that may be related to the task we utilized. The task we employed was created specifically to examine amygdala reactivity and its functional circuitry. However, the involvement of other areas, such as the insula and orbitofrontal cortex (36) , that are potentially involved in the genetic risk for deficits in emotion processing cannot be specifically investigated by our task. Another limitation to our study is related to the design of the control session of the task, consisting of geometric shapes instead of neutral faces, which raises questions regarding the specificity of the task for emotional processing versus face processing. Although we cannot completely eliminate the possibility that the present results can be interpreted as being related only to face processing, the specific engagement of the limbic system as well as the results from previous studies that employed this same task (e.g., 4 , 5 , 7 , 9 , 10 , 37) suggest that the task is capable of examining not only face processing but also the circuit related to threatening stimuli.
Notwithstanding several limitations, our data suggest that the pathophysiological mechanism underlying the inability of schizophrenia patients to normally process fearful and angry facial representations is influenced by treatment and is unlikely related to genetic risk for schizophrenia.
1. LeDoux J: The amygdala. Curr Biol 2007; 17:R868–R874Google Scholar
2. Pinkham AE, Gur RE, Gur RC: Affect recognition deficits in schizophrenia: neural substrates and psychopharmacological implications. Expert Rev Neurother 2007; 7:807–816Google Scholar
3. Brunet-Gouet E, Decety J: Social brain dysfunctions in schizophrenia: a review of neuroimaging studies. Psychiatry Res 2006; 148:75–92Google Scholar
4. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR: Serotonin transporter genetic variation and the response of the human amygdala. Science 2002; 297:400–403Google Scholar
5. Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF, Weinberger DR: A susceptibility gene for affective disorders and the response of the human amygdala. Arch Gen Psychiatry 2005; 62:146–152Google Scholar
6. Heinz A, Braus DF, Smolka MN, Wrase J, Puls I, Hermann D, Klein S, Grüsser SM, Flor H, Schumann G, Mann K, Büchel C: Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat Neurosci 2005; 8:20–21Google Scholar
7. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR: 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 2005; 8:828–834Google Scholar
8. Smolka MN, Schumann G, Wrase J, Grusser SM, Flor H, Mann K, Braus DF, Goldman D, Büchel C, Heinz A: Catechol- O -methyltransferase Val 158 Met genotype affects processing of emotional stimuli in the amygdala and prefrontal cortex. J Neurosci 2005; 25:836–842 Google Scholar
9. Drabant EM, Hariri AR, Meyer-Lindenberg A, Munoz KE, Mattay VS, Kolachana BS, Egan MF, Weinberger DR: Catechol- O -methyltransferase Val 158 Met genotype and neural mechanisms related to affective arousal and regulation. Arch Gen Psychiatry 2006; 63:1396–1406 Google Scholar
10. Meyer-Lindenberg A, Buckholtz JW, Kolachana B, Hariri AR, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Mattay V, Weinberger DR: Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc Natl Acad Sci U S A 2006; 103:6269–6274Google Scholar
11. Toomey R, Seidman LJ, Lyons MJ, Faraone SV, Tsuang MT: Poor perception of nonverbal social-emotional cues in relatives of schizophrenic patients. Schizophr Res 1999; 40:121–130Google Scholar
12. Bolte S, Poustka F: The recognition of facial affect in autistic and schizophrenic subjects and their first-degree relatives. Psychol Med 2003; 33:907–915Google Scholar
13. Gur RE, Nimgaonkar VL, Almasy L, Calkins ME, Ragland JD, Pogue-Geile MF, Kanes S, Blangero J, Gur RC: Neurocognitive endophenotypes in a multiplex multigenerational family study of schizophrenia. Am J Psychiatry 2007; 164:813–819Google Scholar
14. Edwards J, Jackson HJ, Pattison PE: Emotion recognition via facial expression and affective prosody in schizophrenia: a methodological review. Clin Psychol Rev 2002; 22:789–832Google Scholar
15. Goldberg TE, Torrey EF, Gold JM, Ragland JD, Bigelow LB, Weinberger DR: Learning and memory in monozygotic twins discordant for schizophrenia. Psychol Med 1993; 23:71–85Google Scholar
16. Goldberg TE, Torrey EF, Gold JM, Bigelow LB, Ragland RD, Taylor E, Weinberger DR: Genetic risk of neuropsychological impairment in schizophrenia: a study of monozygotic twins discordant and concordant for the disorder. Schizophr Res 1995; 17:77–84Google Scholar
17. Cannon TD, Huttunen MO, Lonnqvist J, Tuulio-Henriksson A, Pirkola T, Glahn D, Finkelstein J, Hietanen M, Kaprio J, Koskenvuo M: The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. Am J Hum Genet 2000; 67:369–382Google Scholar
18. Aleman A, Kahn RS: Strange feelings: Do amygdala abnormalities dysregulate the emotional brain in schizophrenia? Prog Neurobiol 2005; 77:283–298Google Scholar
19. Braff DL, Freedman R, Schork NJ, Gottesman II: Deconstructing schizophrenia: an overview of the use of endophenotypes in order to understand a complex disorder. Schizophr Bull 2007; 33:21–32Google Scholar
20. Callicott JH, Weinberger DR: Brain imaging as an approach to phenotype characterization for genetic studies of schizophrenia. Methods Mol Med 2003; 77:227–247Google Scholar
21. Callicott JH, Egan MF, Mattay VS, Bertolino A, Bone AD, Verchinksi B, Weinberger DR: Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am J Psychiatry 2003; 160:709–719Google Scholar
22. Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR: Effect of COMT Val 108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A 2001; 98:6917–6922 Google Scholar
23. Woods SW: Chlorpromazine equivalent doses for the newer atypical antipsychotics. J Clin Psychiatry 2003; 64:663–667Google Scholar
24. Serra M, Salgado-Pineda P, Delaveau P, Fakra E, Gasto C, Blin O: Effects of antidepressant drugs on emotion. Clin Neuropharmacol 2006; 29:170–185Google Scholar
25. Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276Google Scholar
26. Ekman P, Friesen WV: Pictures of Facial Affect. Palo Alto, Calif, Consulting Psychologists Press, 1976Google Scholar
27. Fisher RA: On the “probable error” of a coefficient of correlation deduced from a small sample. Metron 1921; 1:3–32Google Scholar
28. Canli T, Desmond JE, Zhao Z, Gabrieli JD: Sex differences in the neural basis of emotional memories. Proc Natl Acad Sci U S A 2002; 99:10789–10794Google Scholar
29. Canli T, Zhao Z, Brewer J, Gabrieli JD, Cahill L: Event-related activation in the human amygdala associates with later memory for individual emotional experience. J Neurosci 2000; 20:RC99Google Scholar
30. Phelps EA, O’Connor KJ, Gatenby JC, Gore JC, Grillon CD, Davis M: Activation of the left amygdala to a cognitive representation of fear. Nat Neurosci 2001; 4:437–441Google Scholar
31. Phelps EA, Delgado MR, Nearing KI, LeDoux JE: Extinction learning in humans: role of the amygdala and vmPFC. Neuron 2004; 43:897–905Google Scholar
32. Habel U, Klein M, Shah NJ, Toni I, Zilles K, Falkai P, Schneider F: Genetic load on amygdala hypofunction during sadness in nonaffected brothers of schizophrenia patients. Am J Psychiatry 2004; 161:1806–1813Google Scholar
33. Kosaka H, Omori M, Murata T, Iidaka T, Yamada H, Okada T, Takahashi T, Sadato N, Itoh H, Yonekura Y, Wada Y: Differential amygdala response during facial recognition in patients with schizophrenia: an fMRI study. Schizophr Res 2002; 57:87–95Google Scholar
34. Holt DJ, Kunkel L, Weiss AP, Goff DC, Wright CI, Shin LM, Rauch SL, Hootnick J, Heckers S: Increased medial temporal lobe activation during the passive viewing of emotional and neutral facial expressions in schizophrenia. Schizophr Res 2006; 82:153–162Google Scholar
35. Russell TA, Reynaud E, Kucharska-Pietura K, Ecker C, Benson PJ, Zelaya F, Giampietro V, Brammer M, David A, Phillips ML: Neural responses to dynamic expressions of fear in schizophrenia. Neuropsychologia 2007; 45:107–123Google Scholar
36. Phillips ML, Drevets WC, Rauch SL, Lane R: Neurobiology of emotion perception, II: implications for major psychiatric disorders. Biol Psychiatry 2003; 54:515–528Google Scholar
37. Buckholtz JW, Callicott JH, Kolachana B, Hariri AR, Goldberg TE, Genderson M, Egan MF, Mattay VS, Weinberger DR, Meyer-Lindenberg A: Genetic variation in MAO-A modulates ventromedial prefrontal circuitry mediating individual differences in human personality. Mol Psychiatry 2008; 13:313–324Google Scholar
38. Wechsler D: Wechsler Adult Intelligence Scale—Revised. San Antonio, Tex, Psychological Corporation, 1981Google Scholar
39. Oldfield RC: The assessment and analysis of handedness: The Edinburgh Inventory. Neuropsychologia 1971; 9:97–113Google Scholar

