
Lower Serotonin Transporter Binding Potential in the Human Brain During Major Depressive Episodes
Abstract
OBJECTIVE: CSF analysis, neuroendocrine challenges, serotonin depletion studies, and treatment studies implicate the serotonergic system in the pathophysiology of major depressive disorder. On the basis of postmortem and imaging studies, the authors hypothesized that subjects with major depressive disorder in a major depressive episode have fewer serotonin transporter sites, compared with healthy subjects. METHOD: Serotonin transporter binding potential (f1Bmax/Kd) was determined using positron emission tomography with [11C]McN 5652 in six brain regions in 25 medication-free subjects with DSM-IV major depressive disorder during a major depressive episode and in 43 healthy volunteer comparison subjects. All subjects had arterial lines placed to determine metabolite-corrected arterial input functions. RESULTS: Serotonin transporter binding potential differed significantly by brain region and group. Post hoc analysis revealed lower binding potential in subjects with major depressive disorder, relative to the comparison subjects, in the amygdala and midbrain. The lower binding potential was more pronounced in the depressed subjects who had never received antidepressants. No correlation was found between binding potential in the midbrain and severity of depression or number of days without medication. Binding potential did not differ between suicide attempters and nonattempters. CONCLUSIONS: Subjects in a major depressive episode have lower serotonin transporter binding potential in the amygdala and midbrain, compared to healthy subjects.
Serotonin (5-HT) transmission appears altered in major depressive disorder (1–3). The serotonin transporter plays a critical role in 5-HT transmission by terminating the action of 5-HT by reuptake into presynaptic neurons (4). Serotonin transporter binding is localized to serotonergic neurons (5), and it can be regulated by drugs and by cellular mechanisms (6, 7). Serotonin transporter has been implicated in the pathophysiology of major depressive disorder (8) and is the target of most current antidepressants (5).
Findings from postmortem, lesion, and brain imaging studies have suggested two main anatomical circuits involved in mood regulation (9–11). The first is a limbic-thalamic-cortical circuit that includes the amygdala, medial-dorsal nucleus of the thalamus, and ventrolateral prefrontal cortex. The second is a limbic-striatal-pallidal-thalamic-cortical circuit. Mood disorders may occur if there is a disruption in the functioning of a component of either circuit, both of which receive serotonergic innervation.
In addition to these circuits, the hippocampus and anterior cingulate also have serotonergic abnormalities in major depressive disorder. Female subjects with remitted depression have smaller hippocampal volumes (12), and smaller hippocampal volume is negatively correlated with the duration of lifetime depression (13) and is perhaps related to lower postmortem levels of serotonin transporter (14). The anterior cingulate is implicated in the pathophysiology of major depressive disorder, and there are reports of lower blood flow and metabolism (15–18), smaller volume (18), and lower postmortem serotonin transporter binding (19) for this region. The dorsal and median raphe nuclei supply all the serotonergic projections to the forebrain. An in vivo single photon emission computerized tomography study demonstrated 19% lower [123I]β-CIT serotonin transporter binding in the midbrain of subjects with major depressive disorder, compared with healthy subjects (20).
We used positron emission tomography (PET) with the radiotracer [11C](+)-6β-(4-methylthiophenyl)-1,2,3,5,6α,10β-hexahydropyrrolo[2,1-a]isoquinoline ([11C]McN 5652) to examine serotonin transporter binding potential in brain regions that have quantifiable serotonin transporter, are implicated in the circuits proposed, and have 5-HT abnormalities—the amygdala, hippocampus, thalamus, putamen, anterior cingulate cortex, and midbrain. Healthy volunteer subjects were compared with medication-free subjects with major depressive disorder during a major depressive episode. We hypothesized that lower [11C]McN 5652 binding would be found in the selected regions in subjects with major depressive disorder. [11C]McN 5652 binding in the prefrontal cortex is too low to quantify reliably (21). We also explored the relationship between [11C]McN 5652 binding and previous exposure to antidepressants (22), suicide attempt status, and clinical measures of depression severity.
Method
Subjects
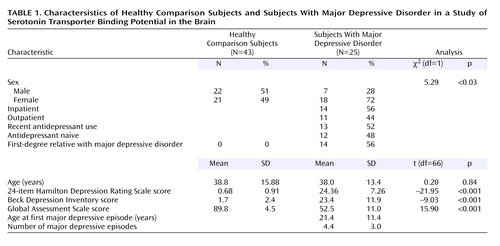
Twenty-five subjects who met the DSM-IV criteria for a current major depressive episode and 43 healthy comparison subjects were included in the study. Their demographic and clinical characteristics are summarized Table 1. Inclusion criteria for subjects with major depressive disorder were 1) age 18–65 years; 2) fulfillment of the DSM-IV criteria for major depressive episode; 3) absence of any psychotropic medication use for at least 2 weeks before the PET scan (6 weeks for fluoxetine, 4 weeks for oral neuroleptics), except for benzodiazepines, which were discontinued 3 days before the scan; 4) no current or lifetime history of alcohol or other drug abuse or dependence; 5) absence of lifetime exposure to 3,4-methylenedioxymethamphetamine; 6) absence of significant current medical conditions; 7) absence of pregnancy; and 8) capacity to provide informed consent. The inclusion criteria for the comparison subjects were similar. In addition, the comparison subjects were required to have no psychiatric history and no history of a mood or psychotic disorder in their first-degree relatives. All subjects gave written informed consent after explanation of the study. The Beck Depression Inventory (BDI) (23), Hamilton Depression Rating Scale (HAM-D) (24), and Global Assessment Scale (GAS) (25) were used to assess subjective and objective depression severity and functional impairment, respectively.
Seven depressed subjects met the criteria for melancholia, four were experiencing their first episode of major depression, and three had too many lifetime episodes to count reliably. Nine depressed subjects had made suicide attempts (mean=1.9 attempts, SD=1.1, range=1–4). Comorbid disorders included posttraumatic stress disorder (N=4), panic disorder (N=7), dysthymia (N=5), social phobia (N=4), generalized anxiety disorder (N=2), binge eating disorder (N=1), and simple phobia (N=1). Twelve subjects had never received antidepressant medications. The mean medication-free interval (excluding benzodiazepines) in the remaining subjects was 26.5 days (SD=13.2, median=21, range=14–58). The subjects who were not medication naive had received medication between 14 and 30 days before the study and had shown no clinical response; they agreed to have their medications tapered and discontinued for the study.
Radiochemistry
[11C]McN 5652 was produced as previously described (26). The injected radioactivity of [11C]McN 5652 differed between the comparison subjects (mean=11.9 mCi, SD=4.3) and the subjects with major depressive disorder (mean=14.3 mCi, SD=3.9) (t=–2.29, df=66, p<0.03), but the injected mass did not differ between groups (comparison subjects: mean=82.2 μmol, SD=62.1; subjects with major depressive disorder: mean=107.8 μmol, SD=156.6) (t=–0.95, df=66, p=0.35).
Image Analysis and Modeling
PET and magnetic resonance imaging data acquisition, analysis, and measurement of arterial input indices were previously described (21). After a 10-minute transmission scan, [11C]McN 5652 was injected intravenously, and emission data were acquired for 130 minutes. After radiotracer injection, 12 arterial samples were collected at 10-second intervals over 2 minutes, the next six samples were collected at 20-second intervals over 2 minutes, and the remaining 13 samples were collected at longer intervals, for a total of 31 samples. The first 18 samples were collected with an automated sampling system, and the remaining samples were manually drawn. Six samples (collected at 2, 20, 50, 80, 110, and 130 minutes) were further processed by high-pressure liquid chromatography to measure the fraction of plasma activity representing unmetabolized parent compound. Regions of interest were traced by using brain atlases (27, 28) and published reports (29, 30). A neuroanatomist (V.A.) verified the regions of interest. Derivation of [11C]McN 5652 regional distribution volume (VT) was performed with likelihood estimation in a graphical approach (31–33). VT is the sum of the specific (V3) and nondisplaceable (free plus nonspecific binding) (V2) distribution volumes. Binding potential (BP′) was calculated as follows: VT – V2, equivalent to f1Bmax/Kd, where f1 is the free concentration of the radiotracer in plasma, Bmax is the maximum number of binding sites, and Kd is the dissociation constant. V2 was measured in a 12.1-cm3 (± 1.5 cm3) sample of the cerebellum.
Statistical Analysis
Data were analyzed by using linear mixed-effects models, with region and diagnostic group as fixed effects and subject as the random effect. To stabilize variance and satisfy modeling assumptions, natural log-transformed data were used in the analysis after a quantity was added to all measures to ensure positive values. The reported p values were not adjusted for multiple comparisons.
Results
The group with major depressive episode included a greater proportion of female subjects, relative to the comparison group (Table 1), but no effect of sex was found in the mixed model (F=0.26, df=1, 63, p=0.62). Across the six regions of interest, BP′ differed significantly by diagnostic group (F=2.82, df=5, 330, p<0.02). In post hoc tests examining the effect of diagnostic group on BP′ in each region in the mixed model, significantly lower BP′ was found in the amygdala (t=–2.29, df=329, p<0.03) and midbrain (t=–2.22, df=329, p<0.03) in the subjects with major depressive disorder, relative to the comparison subjects.
Effect of Previous Exposure to Antidepressants
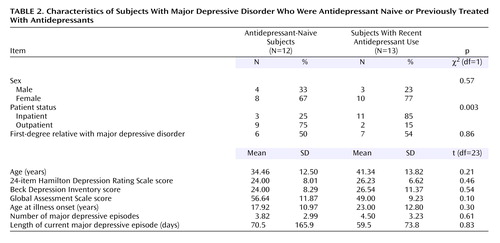
Selective serotonin reuptake inhibitors are cleared from the brain about 2 weeks after discontinuation (34), and therefore, residual drug should not occupy the serotonin transporters after that period. However, medication may have long-term effects such as suppression of gene expression (35), although this possibility is controversial (22, 36). To rule out effects of prior treatment with antidepressants, data for 13 subjects with major depressive disorder who had been medication free for at least 14 days were compared to data for 12 subjects who had never received antidepressants and to data for the comparison subjects. We found a significant effect of region and medication status (F=2.23, df=10, 325, p<0.02) (Figure 1). Post hoc testing revealed significantly lower BP′ in the amygdala (t=–2.93, df=323, p=0.003) (Figure 2) and midbrain (t=–2.37, df=323, p<0.02) for the antidepressant-naive subjects, relative to the comparison subjects. When the data for the comparison subjects were excluded from the analysis, there was no difference in BP′ between the antidepressant-naive and recently medicated groups across all brain regions (F=0.71, df=1, 23, p=0.41), although the interaction of region and medication status approached significance (F=2.24, df=5, 115, p=0.055). The 12 antidepressant-naive subjects did not differ from the 13 recently treated subjects in demographic and clinical characteristics, except that the recently treated subjects were more likely to be inpatients (Table 2). No correlation was found between the time since discontinuation of antidepressants and BP′ in any region of interest (data not shown), although the previously medicated subjects had a median time of only 21 days since discontinuation of antidepressants.
Effect of Suicide Attempt Status
A group-by-region interaction was found in the comparison of BP′ in the subjects with major depressive disorder who had a history of at least one suicide attempt, the subjects with major depressive disorder who had never attempted suicide, and the comparison subjects (F=1.12, df=10, 325, p<0.05). Post hoc analysis revealed that this difference was due to differences in BP′ in the amygdala and midbrain between the comparison subjects and the subjects with major depressive disorder who had never attempted suicide. No differences in BP′ were found between the suicide attempters and nonattempters or between the attempters and the comparison subjects (data not shown).
Correlation Between BP′ and Depression Severity
No significant correlation was found between the BP′ in any region of interest and the measures of depression severity, including the BDI and HAM-D scores, age at first major depressive episode, number of previous major depressive episodes, or length of current major depressive episode.
Discussion
We found lower midbrain and amygdala [11C]McN 5652 BP′ in major depressive disorder and found that this effect was more pronounced in depressed subjects who had never received antidepressants. There was no correlation between BP′ and depression severity. There was no difference in BP′ between the major depressive disorder subjects with a history of suicide attempt and those with no history of suicide attempt.
Comparison to Previous In Vivo Imaging Studies
Our finding of a 20% lower BP′ in the midbrain in subjects with major depressive disorder, compared to healthy volunteers, is in agreement with that of Malison et al. (20), who found a 19% lower [123I]β-CIT binding in subjects with major depressive disorder, but is at odds with the findings of Ichimiya et al. (37), who reported no difference in midbrain [11C]McN 5652 V3′′ (V3′′ is defined later in the Discussion section) and higher V3′′ in the thalamus of depressed subjects. It should be noted that the study by Ichimiya et al. 1) included euthymic subjects; 2) used data from 90-minute scans; 3) used age as a covariate (we could detect no age-related changes in BP′ in the midbrain); 4) used the graphical method of Logan, which is reported to have noise-dependent bias (38); and 5) had a smaller number of depressed subjects (N=13). Our data are also consistent with the results of a previous PET study with the transporter radioligand [11C] N,N-dimethyl-2-(2-amino-4-cyanophenylthio)benzylamine ([11C]DASB) in which no difference in striatal V3′′ was found between depressed subjects and healthy comparison subjects (39). We found a 20% lower BP′ in the amygdala in subjects with major depressive disorder, compared to healthy subjects; to our knowledge, this finding has not been reported previously.
Interpretation of Findings
We interpret the lower [11C]McN 5652 BP′ (f1Bmax/Kd) in the subjects with major depressive disorder to reflect lower Bmax, or lower total number of available serotonin transporter binding sites. Several outcome measures are used in PET studies, including BP (Bmax/Kd=VT – V2/f1), BP′ (f1Bmax/Kd=VT – V2), and V3′′ (f2Bmax/Kd=BP′/V2), where VT is the total volume of distribution in an region of interest, V2 is a measure of the free and nonspecifically bound tracer, f1 is the free concentration of the radiotracer in plasma, and f2 is the free concentration of the radiotracer in the cerebrospinal fluid. Ideally we would measure BP, but f1 is not measurable with [11C]McN 5652. Our findings were similar when either of the outcome measures that can be determined with [11C]McN 5652 was used, i.e., the principal region-by-diagnosis interaction was significant with BP′ or V3′′ (p<0.05), and the interaction of region and prior antidepressant medication status was significant with V3′′ (p<0.03). We believe the lower BP′ in major depressive disorder is not a consequence of f1, because our findings are consistent with postmortem findings, where f1 is not a factor, and because changes in f1 would result in global differences, not regionally specific ones. Finally, postmortem studies suggested that lower levels of serotonin transporter in subjects with major depressive disorder are not due to changes in receptor affinity (1/d) (14). It is unlikely that the lower BP′ in major depressive disorder could be a consequence of higher synaptic levels of 5-HT, because of the hypothesized serotonin abnormality in depression. Moreover, in postmortem studies that included a preincubation period to promote catabolism and a washout of endogenous 5-HT before adding ligand in vitro, lower serotonin transporter binding was found in major depressive disorder (40–42). These findings appear to rule out elevated intrasynaptic levels of 5-HT as an explanation. Therefore, our finding of lower serotonin transporter BP′ in major depressive disorder is likely a result of lower serotonin transporter Bmax, i.e., fewer serotonin transporters.
There are several possible explanations for the lower serotonin transporter Bmax in major depressive disorder. Lower levels of synaptic 5-HT could facilitate serotonin transporter internalization (6); there could be fewer 5-HT neurons or neuronal processes or decreased expression of serotonin transporter per terminal; or a combination of these factors could be operating. Lower levels of synaptic 5-HT in the raphe nuclei could be related to the presence of the C(-1019)G polymorphism of the promoter region of the 5-HT1A gene that has been shown to be overrepresented in patients with major depressive disorder (43). The C(-1019) allele is part of a 26-base pair imperfect palindrome with less affinity for the nuclear deformed epidermal autoregulatory factor transcriptional repressor, and this lower level of affinity could potentially result in a higher level of expression of the 5-HT1A protein in the raphe nuclei. Because the raphe nuclei 5-HT1A receptors are inhibitory, higher levels of 5-HT1A in the raphe nuclei would result in lower levels of 5-HT release and possible down-regulation of serotonin transporter. Alternatively, lower levels of synaptic 5-HT could be related to having a variant of the tryptophan hydroxylase-2 gene, which has less catalytic activity and is present in 10% of subjects with major depressive disorder (44, 45). Down-regulation of serotonin transporter should be accompanied by reduced mRNA for serotonin transporter. Our previous autoradiography studies suggested that the number of neurons that express serotonin transporter mRNA is reduced by 54% in subjects with major depressive disorder who commit suicide (42), although the number of 5-HT-producing neurons did not appear to be decreased (46).
We hypothesized that a lower level of serotonin transporter in all six regions of interest was necessary for the expression of major depressive disorder, but we found differences in serotonin transporter binding potential only in the midbrain and amygdala. These findings can be interpreted in several ways. First, it is possible that the midbrain and amygdala are the crucial structures in the pathophysiology of depression, and serotonin transporter abnormalities in these regions are sufficient to result in major depressive disorder. Second, the abnormalities in the other regions of interest may not be specific to serotonin transporter but perhaps may involve other measures of serotonergic function. Third, the lower level of serotonin transporter BP′ in the midbrain and amygdala may be secondary to as yet undetermined primary pathological process(es). Finally, the PET ligand we used may not be sensitive enough to detect serotonin transporter abnormalities in the other regions.
For the differences in serotonin transporter BP′ to be limited to the midbrain and amygdala, the abnormality may be confined to a subpopulation of serotonin-transporter-expressing 5-HT neurons within the dorsal raphe nuclei. Less 5-HT input to the amygdala, as suggested by the finding of lower serotonin transporter BP′, may result in increased amygdala activity (47), as serotonin enhances inhibition in the amygdala, presumably through activation of γ-aminobutyric-acid-ergic interneurons (48). Serotonergic projections to the amygdala are implicated in anxiety disorders as well (49). Previous research has suggested an association between hyperactivity in the amygdala and a greater likelihood that sensory or social stimuli are perceived or remembered as emotionally arousing or aversive (50); thus, hyperactivity in the amygdala may contribute to the relationship between adverse childhood experiences and adult mood disorders.
 |
 |
Received June 2, 2004; revisions received Jan. 11 and March 1, 2005; accepted April 13, 2005. From the Departments of Psychiatry and Radiology, Columbia University College of Physicians and Surgeons, New York; the Departments of Neuroscience and Analytic Psychopharmacology, New York State Psychiatric Institute; and the Department of Biostatistics, Columbia University School of Public Health, New York. Address correspondence and reprint requests to Dr. Parsey, Department of Neuroscience, New York State Psychiatric Institute, 1051 Riverside Dr., Box 42, New York, NY 10032; [email protected] (e-mail). This article is the second of two on serotonin transporters in the human brain in this issue of the Journal. The first article immediately precedes this one. Supported by NIMH grants P30 MH-46745, MH-40695, MH-40210, and MH-62185, the National Alliance for Research on Schizophrenia and Depression, and the American Foundation for Suicide Prevention. The authors thank the staff of the Brain Imaging Division, Kreitchman PET Center, and Sarina M. Rodrigues, Ph.D., for their assistance.

Figure 1. Serotonin Transporter Binding Potential in Healthy Comparison Subjects and Subjects With Major Depressive Disorder Who Had Never Been Treated With Antidepressants or Who Had Been Recently Treated With Antidepressants a
aSignificant effect of region and group (F=2.23, df=10, 325, p<0.02).
bSignificantly lower binding potential in the depressed subjects who had never received antidepressants, relative to the comparison subjects (t=–2.37, df=323, p<0.02).
cSignificantly lower binding potential in the depressed subjects who had never received antidepressants, relative to the comparison subjects (t=–2.93, df=323, p=0.003).

Figure 2. Serotonin Transporter Binding Potential in the Amygdala in Healthy Comparison Subjects and Subjects With Major Depressive Disorder Who Had Never Been Treated With Antidepressants or Who Had Been Recently Treated With Antidepressantsa
aBars represent mean values.
1. Lucki I: The spectrum of behaviors influenced by serotonin. Biol Psychiatry 1998; 44:151–162Crossref, Medline, Google Scholar
2. Deakin JF: Depression and 5-HT. Int Clin Psychopharmacol 1991; 6(suppl 3):23-28Google Scholar
3. Mann JJ: Role of the serotonergic system in the pathogenesis of major depression and suicidal behavior. Neuropsychopharmacology 1999; 21(2 suppl):99S-105SGoogle Scholar
4. Rudnick G, Clark J: From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta 1993; 1144:249–263Crossref, Medline, Google Scholar
5. Soucy JP, Lafaille F, Lemoine P, Mrini A, Descarries L: Validation of the transporter ligand cyanoimipramine as a marker of serotonin innervation density in brain. J Nucl Med 1994; 35:1822–1830Medline, Google Scholar
6. Ramamoorthy S, Blakely RD: Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science 1999; 285:763–766Crossref, Medline, Google Scholar
7. Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD: Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci 1997; 17:45–57Crossref, Medline, Google Scholar
8. Owens MJ, Nemeroff CB: Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem 1994; 40:288–295Medline, Google Scholar
9. Drevets WC: Functional neuroimaging studies of depression: the anatomy of melancholia. Annu Rev Med 1998; 49:341–361Crossref, Medline, Google Scholar
10. Soares JC, Mann JJ: The functional neuroanatomy of mood disorders. J Psychiatr Res 1997; 31:393–432Crossref, Medline, Google Scholar
11. Mayberg HS: Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci 1997; 9:471–481Crossref, Medline, Google Scholar
12. Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW: Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996; 93:3908–3913Crossref, Medline, Google Scholar
13. Sheline YI, Sanghavi M, Mintun MA, Gado MH: Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 1999; 19:5034–5043Crossref, Medline, Google Scholar
14. Perry EK, Marshall EF, Blessed G, Tomlinson BE, Perry RH: Decreased imipramine binding in the brains of patients with depressive illness. Br J Psychiatry 1983; 142:188–192Crossref, Medline, Google Scholar
15. Baxter LR Jr, Schwartz JM, Phelps ME, Mazziotta JC, Guze BH, Selin CE, Gerner RH, Sumida RM: Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch Gen Psychiatry 1989; 46:243–250Crossref, Medline, Google Scholar
16. Baxter LR Jr, Phelps ME, Mazziotta JC, Schwartz JM, Gerner RH, Selin CE, Sumida RM: Cerebral metabolic rates for glucose in mood disorders: studies with positron emission tomography and fluorodeoxyglucose F 18. Arch Gen Psychiatry 1985; 42:441–447Crossref, Medline, Google Scholar
17. Mann JJ, Malone KM, Diehl DJ, Perel J, Nichols TE, Mintun MA: Positron emission tomographic imaging of serotonin activation effects on prefrontal cortex in healthy volunteers. J Cereb Blood Flow Metab 1996; 16:418–426Crossref, Medline, Google Scholar
18. Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, Raichle ME: Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997; 386:824–827Crossref, Medline, Google Scholar
19. Arango V, Underwood MD, Mann JJ: Serotonin brain circuits involved in major depression and suicide. Prog Brain Res 2002; 136:443–453Crossref, Medline, Google Scholar
20. Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L, Sanacora G, Owens MJ, Nemeroff CB, Rajeevan N, Baldwin RM, Seibyl JP, Innis RB, Charney DS: Reduced brain serotonin transporter availability in major depression as measured by [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry 1998; 44:1090–1098Crossref, Medline, Google Scholar
21. Parsey RV, Kegeles LS, Hwang DR, Simpson N, Abi-Dargham A, Mawlawi O, Slifstein M, Van Heertum RL, Mann JJ, Laruelle M: In vivo quantification of brain serotonin transporters in humans using [11C]McN 5652. J Nucl Med 2000; 41:1465–1477Medline, Google Scholar
22. Blakely RD, Ramamoorth S, Qian Y, Schroeter S, Bradley C: Regulation of Antidepressant-Sensitive Serotonin Transporters. Totowa, NJ, Humana Press, 1997Google Scholar
23. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J: An inventory for measuring depression. Arch Gen Psychiatry 1961; 4:561–571Crossref, Medline, Google Scholar
24. Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–62Crossref, Medline, Google Scholar
25. Endicott J, Spitzer RL, Fleiss JL, Cohen J: The Global Assessment Scale: a procedure for measuring overall severity of psychiatric disturbance. Arch Gen Psychiatry 1976; 33:766–771Crossref, Medline, Google Scholar
26. Frankle WG, Huang Y, Hwang DR, Talbot PS, Slifstein M, Van Heertum R, Abi-Dargham A, Laruelle M: Comparative evaluation of serotonin transporter radioligands 11C-DASB and 11C-McN 5652 in healthy humans. J Nucl Med 2004; 45:682–694Medline, Google Scholar
27. Duvernoy HM: The Human Brain: Surface, Three-Dimensional Sectional Anatomy and MRI. New York, Springer-Verlag Wien, 1991Google Scholar
28. Talairach J, Tournoux P: Co-Planar Stereotaxic Atlas of the Human Brain: Three-Dimensional Proportional System. New York, Thieme Medical, 1988Google Scholar
29. Kates WR, Abrams MT, Kaufmann WE, Breiter SN, Reiss AL: Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatr Res Neuroimaging 1997; 75:31–48Crossref, Medline, Google Scholar
30. Killiany RJ, Moss MB, Nicholson T, Jolesz F, Sandor T: An interactive procedure for extracting features of the brain from magnetic resonance images: the lobes. Hum Brain Mapp 1997; 5:355–363Crossref, Medline, Google Scholar
31. Ogden RT: On estimation of kinetic parameters in graphical analysis of PET imaging data. Stat Med 2003; 22:3557–3568Crossref, Medline, Google Scholar
32. Ogden RT, Parsey RV, Mann JJ: Likelihood approach to parameter estimation in Logan graphical analysis (abstract). Neuroimage 2002; 16:S73Google Scholar
33. Parsey RV, Ogden RT, Mann JJ: Determination of volume of distribution using likelihood estimation in graphical analysis: elimination of estimation bias. J Cereb Blood Flow Metab 2003; 23:1471–1478Crossref, Medline, Google Scholar
34. Strauss WL, Layton ME, Dager SR: Brain elimination half-life of fluvoxamine measured by 19F magnetic resonance spectroscopy. Am J Psychiatry 1998; 155:380–384Link, Google Scholar
35. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, Frazer A: Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci 1999; 19:10494–10501Crossref, Medline, Google Scholar
36. Kovachich GB, Aronson CE, Brunswick DJ: Effect of repeated administration of antidepressants on serotonin uptake sites in limbic and neocortical structures of rat brain determined by quantitative autoradiography. Neuropsychopharmacology 1992; 7:317–324Medline, Google Scholar
37. Ichimiya T, Suhara T, Sudo Y, Okubo Y, Nakayama K, Nankai M, Inoue M, Yasuno F, Takano A, Maeda J, Shibuya H: Serotonin transporter binding in patients with mood disorders: a PET study with [11C](+)McN5652. Biol Psychiatry 2002; 51:715–722Crossref, Medline, Google Scholar
38. Slifstein M, Laruelle M: Effects of statistical noise on graphic analysis of PET neuroreceptor studies. J Nucl Med 2000:2083-2088Google Scholar
39. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K, Houle S: Occupancy of serotonin transporters by paroxetine and citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry 2001; 158:1843–1849Link, Google Scholar
40. Arango V, Underwood MD, Gubbi AV, Mann JJ: Localized alterations in pre- and postsynaptic serotonin binding sites in the ventrolateral prefrontal cortex of suicide victims. Brain Res 1995; 688:121–133Crossref, Medline, Google Scholar
41. Mann JJ, Huang YY, Underwood MD, Kassir SA, Oppenheim S, Kelly TM, Dwork AJ, Arango V: A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry 2000; 57:729–738Crossref, Medline, Google Scholar
42. Arango V, Underwood MD, Boldrini M, Tamir H, Kassir SA, Hsiung S, Chen JJ, Mann JJ: Serotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims. Neuropsychopharmacology 2001; 25:892–903Crossref, Medline, Google Scholar
43. Lemonde S, Turecki G, Bakish D, Du L, Hrdina PD, Bown CD, Sequeira A, Kushwaha N, Morris SJ, Basak A, Ou XM, Albert PR: Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci 2003; 23:8788–8799Crossref, Medline, Google Scholar
44. Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG: Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science 2004; 305:217Crossref, Medline, Google Scholar
45. Zhang X, Gainetdinov RR, Beaulieu JM, Sotnikova TD, Burch LH, Williams RB, Schwartz DA, Krishnan KR, Caron MG: Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron 2005; 45:11–16Crossref, Medline, Google Scholar
46. Underwood MD, Khaibulina AA, Ellis SP, Moran A, Rice PM, Mann JJ, Arango V: Morphometry of the dorsal raphe nucleus serotonergic neurons in suicide victims. Biol Psychiatry 1999; 46:473–483Crossref, Medline, Google Scholar
47. Abercrombie HC, Schaefer SM, Larson CL, Oakes TR, Lindgren KA, Holden JE, Perlman SB, Turski PA, Krahn DD, Benca RM, Davidson RJ: Metabolic rate in the right amygdala predicts negative affect in depressed patients. Neuroreport 1998; 9:3301–3307Crossref, Medline, Google Scholar
48. Stutzmann GE, LeDoux JE: GABAergic antagonists block the inhibitory effects of serotonin in the lateral amygdala: a mechanism for modulation of sensory inputs related to fear conditioning. J Neurosci 1999; 19:RC8Google Scholar
49. Stein DJ, Stahl S: Serotonin and anxiety: current models. Int Clin Psychopharmacol 2000; 15(suppl 2):S1-S6Google Scholar
50. Drevets WC: Neuroimaging abnormalities in the amygdala in mood disorders. Ann NY Acad Sci 2003; 985:420–444Crossref, Medline, Google Scholar

