
Relationship Between Release of Platelet/Endothelial Biomarkers and Plasma Levels of Sertraline and N-Desmethylsertraline in Acute Coronary Syndrome Patients Receiving SSRI Treatment for Depression
Abstract
OBJECTIVE: In a platelet/endothelial biomarker substudy of the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART), the authors sought to determine whether plasma levels of sertraline and its primary metabolite N-desmethylsertraline affect the release of platelet/endothelial biomarkers. METHOD: Fifty-five acute coronary syndrome patients with depression were randomly assigned to receive sertraline (N=23) or placebo (N=32). Twenty-six serial plasma samples collected at week 6 (N=12) and week 16 (N=14) were analyzed. Platelet factor 4 (PF4), β-thromboglobulin (β-TG), platelet/endothelial cell adhesion molecule 1 (PECAM-1), P-selectin, thromboxane B2 (TxB2), prostacyclin (6-keto-PGF1α), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin were measured by enzyme-linked immunosorbent assay. Concentrations of sertraline and N-desmethylsertraline were determined by liquid chromatography with fluorescence detection in autologous samples. RESULTS: Strong, mostly time-dependent negative correlations were found for the plasma levels of sertraline and N-desmethylsertraline with PF4 (week 6: r=–0.69 and –0.33, respectively; week 16: r=–0.63 for both), β-TG (week 6: r=–0.43 and –0.29; week 16: r=–0.66 and –0.57), PECAM-1 (week 6: r=–0.82 and –0.49; week 16: r=–0.60 for both), P-selectin (week 6: r=–0.82 and –0.49; week 16: r=–0.73 and –0.43), and TxB2 (week 6: r=–0.66 and –0.59; and week 16: r=–0.64 and –0.41). Regression analysis revealed some borderline correlations for endothelial markers such as 6-keto- PGF1α and E-selectin and a positive correlation for VCAM-1. CONCLUSIONS: This is the first documented evidence that plasma release of platelet/endothelial biomarkers is directly related to the levels of sertraline and N-desmethylsertraline in acute coronary syndrome patients receiving SSRI treatment for depression. The clinical significance of these findings should be assessed in the setting of a randomized clinical trial.
Clinical depression has been identified as an independent risk factor for increased mortality in patients after acute coronary syndromes (1, 2). Selective serotonin reuptake inhibitors (SSRIs) are established agents for treatment of mood disorders including depression. Many investigators have linked possible mortality benefits of SSRIs with their antidepressant properties, regulation of sympathetic and parasympathetic balance, and modulation of vascular tone via dopamine and norepinephrine blockade (3–5). Recently discovered platelet inhibition by SSRIs may represent an independent avenue of pharmacological effects responsible for the benefit of these drugs beyond their effects on depression in patients after acute vascular syndromes such as myocardial infarction and ischemic stroke (6–8).
Sertraline hydrochloride (1S,4S-N-methyl-4-)3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-naphthylamine) is a third-generation antidepressant and a potent SSRI. Sertraline is one of three SSRIs (the others are fluvoxamine and paroxetine) that does not have a neurologically active metabolite. Its major liver breakdown product, N-desmethylsertraline, is a result of sertraline demethylation and is believed to be clinically inactive (9). The Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) was a multicenter, randomized, double-blind, parallel, placebo-controlled trial designed to investigate the safety, tolerability, and antidepressant efficacy of 24 weeks of sertraline treatment for depression in acute coronary syndrome patients (10). Considering that the relative effects of sertraline and N-desmethylsertraline on platelets in the clinical setting are not known, a platelet/endothelial biomarker substudy was implemented in the frame of the index trial (11). Given that acute coronary syndrome patients are usually extensively treated with antiplatelet agents, the potential mortality benefits of SSRIs could be related to the antidepressant properties per se, and improved quality of life, rather than to the direct antiplatelet efficacy. We have shown that sertraline and probably other SSRIs improve platelet markers even in the presence of aspirin (12). In order to establish the relative role of sertraline or N-desmethylsertraline on platelet function and whether that effect is dose dependent, plasma drug levels were measured simultaneously with biomarkers. Thus, the purpose of the present study was to determine whether levels of sertraline and N-desmethylsertraline in autologous plasma samples would affect release of platelet/endothelial markers in patients enrolled in the SADHART trial and whether that release would be dose dependent.
Method
All studies were approved by the investigational review boards of the enrolling sites. Data from the SADHART platelet substudy were analyzed. Platelet/endothelial biomarkers were measured in 165 plasma samples (obtained at baseline and 6 and 16 weeks after treatment group assignment) from 55 patients (23 receiving sertraline, and 32 receiving placebo). Eleven plasma levels of sertraline and N-desmethylsertraline were serially measured by high-performance liquid chromatography with fluorescence probe in 14 of the sertraline-treated patients at week 6 (N=12) and week 16 (N=14) if the autologous platelet-poor plasma samples were still available after measure of biomarkers. Patients with a history of bleeding diathesis, stroke in the preceding 3 months, drug or alcohol abuse, prothrombin time 1.5 times greater than control level, platelet count <100,000/mm3, hematocrit <25%, or creatinine >4.0 mg/dl were excluded. Among the SADHART participants, 89% of the patients received aspirin, 17% received clopidogrel or ticlopidine, and 29% received warfarin sodium (10). In the SADHART cohort analyzed in the present study (11), all patients except one received at least 81 mg/day of aspirin, 20% were treated with clopidogrel, and 26% received warfarin sodium at the manifestation of the acute coronary event. At the time of obtaining blood samples for the index study, all patients were being treated with aspirin, and none was receiving clopidogrel or oral anticoagulants. Patients were evaluated with the structured NIMH Diagnostic Interview Schedule for major depression and completed the self-rated Beck Depression Inventory and Clinical Global Impression improvement scale. All patients enrolled in the study met DSM-IV criteria for major depression. The diagnosis of major depressive disorder was confirmed by a site psychiatrist, and the acute coronary syndrome diagnosis was confirmed by an attending cardiologist.
Samples
Blood samples were obtained with a 19-gauge needle by direct venipuncture and drawn into two 7-ml Vacutainer tubes at room temperature containing 3.8% trisodium citrate. The Vacutainer tube was filled to capacity and gently inverted three to five times to ensure complete mixing of the anticoagulant. The blood-citrate mixture was centrifuged at 3000 g for 10 minutes. The resulting platelet-poor plasma was frozen at –80°C and kept in batches for sertraline levels and platelet/endothelial biomarker assays.
Biomarkers
Platelet factor 4 (PF4), β-thromboglobulin (β-TG) (Diagnostica Stago, Lyon, France), platelet/endothelial cell adhesion molecule-1 (PECAM-1), P-selectin, vascular cell adhesion molecule 1 (VCAM-1), E-selectin (R&D Systems, Minneapolis), thromboxane B2 (TxB2), and prostacyclin (6-keto-PGF1α) (Cayman Chemical, Ann Arbor, Mich.) were measured by enzyme-linked immunosorbent assays in duplicates according to international standards.
Drug Levels
Plasma sertraline and its metabolite N-desmethylsertraline were quantified by using high-performance liquid chromatography with fluorescence detection following precolumn derivatization with dansyl chloride. The method is based upon a previously published procedure for fluoxetine and norfluoxetine with some minor changes (13). In short, 1 ml of plasma containing 150 ng of the bromo analog of sertraline as the internal standard is rendered alkaline with carbonate buffer and extracted with 6 ml of 20% ethyl acetate in n-heptane. After mixing and centrifuging, the organic phase is back extracted with 150 μl of diluted HCl. Following mixing and centrifuging, the organic phase was aspirated and the aqueous phase evaporated to dryness. The residue was derivatized with 1% dansyl chloride for 45 minutes, evaporated to dryness, and reconstituted with mobile phase. Chromatography was carried out by using a Supelco LC-18 5 u 250 mm × 4.6 mm i.d. column and a mobile phase of 15:10:75 (phosphate buffer: methanol: acetonitrile) at a flow rate of 2.0 ml/minute. The eluent was monitored using a fluorescence detector with excitation wavelength set at 235 nm with a cutoff filter at 470 nm. The calibration curve was linear between 800 and 10 ng/ml, and the minimum quantifiable limit was about 5 ng/ml. Total chromatographic analysis time was less than 15 minutes with a within-day imprecision error not exceeding 4.3% and 3.8% for sertraline and N-desmethylsertraline, respectively.
Statistics
A simple linear regression analysis was performed to identify the relationship between individual plasma concentrations of platelet/endothelial biomarkers and the level of sertraline and its major metabolite. Correlations >0.50 were considered significant.
To appraise the hypotheses of interest relates to whether over time there are differences in the biomarkers. Within and between sertraline and placebo groups the following issues were addressed. The comparability of the treatment groups for the baseline characteristics were compared by using t tests. Repeated measures analyses, in which the repeated measures were week 6 and week 16, were utilized to determine whether there were biomarker level differences between treatment groups. The mixed model incorporated the repeated measure week, baseline as a covariate, and patient as a random effect. For plasma biomarkers, Y, the following model was used:
Y=b0 + b1 baseline plasma biomarker + b2 Z + b3 week + b4 week-by-treatment + patient + error.
Here Z is an indicator variable taking the value 1 for sertraline and zero otherwise and week-by-treatment is the interaction of week and treatment factors. The SAS procedure Proc MIXED was applied to allow inclusion of subjects with data missing at one of the weeks. To test for differences between treatment groups, the null hypothesis (b2=0) was tested.
In addition, a paired t was used to test whether sertraline and N-desmethylsertraline plasma levels among patients in the sertraline group were the same at week 6 and week 16.
Finally, to test if the plasma biomarker value over time is related to plasma level (sertraline and N-desmethylsertraline), the following repeated measure model was used for subjects receiving sertraline.
Y=b0 + b1 Baseline plasma biomarker + b2 plasma level + b3 week + b4 plasma level-by-week + patient + error.
Results
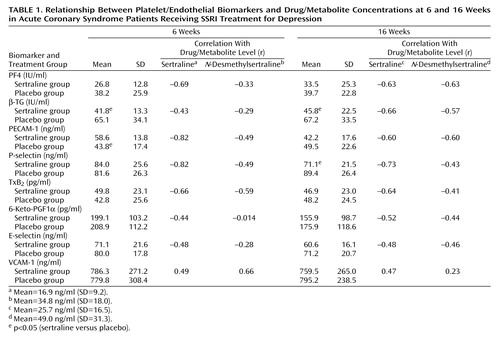
Combined data from all biomarkers/drug level measurements dependent on the length of treatment are presented in Table 1, and representative individual tracings for platelet factor 4 are exhibited in Figure 1.
Strong, mostly time-dependent negative correlations between the plasma levels of sertraline and N-desmethylsertraline were found for predominantly platelet-released markers such as PF4, β-TG, PECAM-1, P-selectin, and TxB2. Regression analysis reveals a borderline correlation for endothelial markers such as 6-keto-PGF1α, and E-selectin, and a surprising positive correlation for VCAM-1 (individual data not shown).
There were no significant differences in plasma biomarker levels between the two treatment groups at baseline. Of the 23 patients treated with sertraline, only 14 had sertraline plasma level data at both weeks. For these subjects, there was a statistical difference between the levels in these two measures, with the average sertraline plasma level higher in the later week. Fifteen subjects had N-desmethylsertraline plasma level data for both weeks, and the difference was not significant over time.
The index data showed significant between-group differences in mean β-TG and P-selectin levels, with the sertraline group lower than the placebo group. There was a significant time effect for P-selectin, with week 16 lower than week 6. There was also significant treatment-by-time interaction for PECAM-1, with the placebo means for weeks 6 and 16 not significantly different from each other while week 16 was significantly lower than week 6 for sertraline, i.e., the difference between plasma levels in weeks 6 and 16 was not independent of group assignment.
There was a significant regression on sertraline plasma levels for seven of the eight biomarker variables. The slopes were negative ones for all biomarkers but VCAM-1. The statistical analysis of the full model failed to converge for PGF1α, but removing the interaction effect from the model resulted in a significant negative slope for the sertraline plasma level. The N-desmethylsertraline plasma level showed a significant regression for the four variables—PECAM-1, PF4, P-selectin, and VCAM-1. All but VCAM-1 have a negative slope. In addition for sertraline plasma levels, P-selectin and VCAM-1 had a significant interaction effect, as did thromboxane for N-desmethylsertraline. For both measures of plasma level, the slope of week 6 was more negative than the slope of week 16.
Discussion
The data from the present study show that plasma levels of sertraline, and its major metabolite N-desmethylsertraline, are strongly negatively correlated with the release of platelet/endothelial biomarkers. This study provides evidence that treatment with sertraline in therapeutic concentrations indeed exhibits antiplatelet and endothelium-protective properties. The difference and added strength is the incorporation of the baseline variables in the model as a covariate and adjustment of week 6 and week 16 data in a single model.
While clinical relevance of these findings is yet unclear, there are several important implications regarding potential vascular survival benefits of SSRIs through their actions not only as antidepressants but also as platelet inhibitors. Platelet inhibition by SSRIs may represent an independent pharmacological action responsible for the ultimate success of this class of agents treating not only depression but also vascular thrombotic events. Indeed, future strategies in patients after vascular events must consider safe approaches to protect platelets from extensive activation. Platelet function is clearly heterogeneous in patients who experience a myocardial infarction (14) or stroke (15) and undergoes dramatic phasic changes after reperfusion therapy (16). Moreover, severe bleeding complications after uniform aggressive strategies with the intravenous GP IIb/IIIa inhibitors could substantially further impair decreased or normal baseline platelet characteristics observed in some patients after acute vascular events.
The postulate that clinical depression is associated with the changes of platelet function and that SSRIs favorably affect platelets is not new. It has been consistently observed that patients with major depression exhibit significant differences in the number of binding sites for various compounds, including imipramine (17), paroxetine (18), and inositol (19) on the platelet surface than comparison subjects. Platelet monoamine oxidase activity is significantly elevated in depressed patients (20, 21), especially in women (22). However, the ability of SSRIs to modulate platelet function is not clear. One study revealed that treatment with sertraline for 8 weeks resulted in a significant reduction of serotonin-mediated platelet aggregation relative to the control values (23). There are also few sporadic observations that SSRIs, and especially fluoxetine, may be responsible for the profound inhibition of platelet aggregation induced by ADP, epinephrine, ristocetin, arachidonic acid, and collagen, resulting in mild bleeding complications followed by rapid normalization of platelet function after the withdrawal of the drug (24, 25). These side effects may be at least in part related to the physiology of serotonin pool distribution in humans. Indeed, the majority of the body’s serotonin is found outside the central nervous system (26). For instance, excessive transcardiac accumulation of serotonin has been demonstrated in patients as chronic stable angina converts to an unstable coronary syndrome (27). Thus, SSRI treatment may result in redistribution of peripheral serotonin by presumably blocking platelet uptake or impacting pulmonary endothelial metabolism of serotonin pool. However, another study of depressed patients treated with fluoxetine or paroxetine found no differences in platelet aggregation after 2 and 4 weeks of treatment when compared with baseline values (28). It is interesting that Alderman et al. (28) in their conclusions acknowledge the possibility that abnormal platelet function is more likely to occur when high doses of SSRIs are administered, which is in agreement with the time- and dose-dependent inhibition of platelet release reactions found in the present study.
Realizing that SSRI therapy affects coagulation biomarkers, we conducted a pilot hemostasis substudy in the frame of our SADHART study and found no changes in coagulation measures after treatment with sertraline (3). A previous report failed to demonstrate any compromised hemostatic function associated with fluoxetine therapy (29), while few random observations have appeared to implicate fluoxetine as a cause of impaired bleedings. Various adverse effects from SSRI use, such as bruising, hematomas, petechiae or purpura, epistaxis (and more rarely intestinal hemorrhage, ocular bleeding, or cerebral hemorrhage) have been anecdotally reported (30), which suggests platelet involvement. However, we are not aware of any published data when biomarker release has been correlated with the plasma SSRI concentrations that link these events in a cause-result relationship. Moreover, the index data are unbiased, since two different laboratories were blinded in the performance of sertraline/N-desmethylsertraline assays and platelet/endothelial measures in the setting of a placebo-controlled, randomized, multicenter clinical trial.
The most unexpected finding of the present study was the fact that N-desmethylsertraline, a neurologically inactive sertraline metabolite, exhibited negative correlations with the release of biomarkers, which match or exceed those of the parent compound. Considering the index results in light of the previously reported antiplatelet properties of sertraline and N-desmethylsertraline in vitro (6), together with their slow absorption and prolonged half-life (9), the platelet inhibition with the metabolite may provide an important clinical benefit to synergically maintain low platelet activity during chronic sertraline administration. This tandem combination may represent a safe and potent alternative or adjunct to currently available antiplatelet agents. Our data are also in agreement with the observation that fluoxetine and norfluoxetine exhibited similar serotonin-blocking effects in rat platelets (31).
The final important issue is that the results of the present study slightly differ from those of the SADHART platelet study per se. Independent from determining plasma levels of sertraline and N-desmethylsertraline, significant differences were observed for the reduction of β-TG (p=0.03) at week 6 and 16, and P-selectin (p=0.04) at week 16 in the sertraline group (11), which is in full agreement with the present report. However, changes in the other platelet-derived biomarkers, so prominent in the index study, such as PF4, PECAM-1, and TxB2 did not favor sertraline (11). This discrepancy is most likely related to the diminished study group size due to insufficient amount of plasma for the drug level tests. Regression analyses differ substantially from the analyses of variance used in the SADHART platelet substudy, therefore precluding certain correlations between the datasets.
There are several limitations of this study. First, the number of subjects was relatively small, and it is difficult to draw definite conclusions. Larger prospective trials are needed to establish the clinical relevance of the described laboratory data. It will be also very useful to examine the properties of the agents using several different types of anticoagulants (such as PPAC, trypsin inhibitor, and trisodium citrate) to explore if the antiplatelet effects are consistent across the other anticoagulants. Another important issue is the need to correlate other established tests measuring platelet anatomy (flow cytometry) and function (aggregometry) with drug levels. In this regard some direct measure of platelet function would have been desirable, while a variety of marker releases do not represent the combined effect on actual platelet function. It would also be important to investigate the influences of intraplatelet calcium concentrations after sertraline and N-desmethylsertraline.
In conclusion, this is the first documented evidence that platelet function is strongly negatively correlated to the plasma levels of sertraline and N-desmethylsertraline, thereby linking survival benefits of SSRI therapy mostly to platelet inhibition rather than endothelial protection in a setting of a major randomized controlled clinical trial. The control for baseline biomarker level differences and the use of placebo to control for the passage of time indicate that the subsequent changes are a true medication effect.
 |
Presented in part at the American Heart Association’s Scientific Sessions 2003, Orlando, Nov. 9–12. Received Jan. 28, 2004; revision received June 2, 2004; accepted June 14, 2004. From Johns Hopkins University; the Analytical Psychopharmacology Laboratories, Nathan S. Kline Institute, Orangeburg, N.Y.; Columbia University, New York; Queen’s University, Kingston, Ont., Canada; and Duke University Medical Center, Durham, N.C. Address correspondence and reprint requests to Dr. Serebruany, HeartDrug Research Laboratories, Johns Hopkins University, 7600 Osler Dr., Suite 307, Towson, MD 21204; [email protected] (e-mail). This study was supported in part by Pfizer (New York). The authors thank the staff of all participating sites whose commitment made this study possible.

Figure 1. Correlation Between Platelet Factor 4 Level and Plasma Levels of Sertraline and N-Desmethylsertraline at 6 and 16 Weeks in Acute Coronary Syndrome Patients Receiving SSRI Treatment for Depression
1. Carney RM, Rich MW, Tevelde A, Saini J, Clark K, Jaffe AS: Major depressive disorders in coronary artery disease. Am J Cardiol 1987; 60:1273–1275Crossref, Medline, Google Scholar
2. Ahern DK, Gorkin L, Anderson JL, Tierney C, Hallstrom A, Ewart C, Capone RJ, Schron E, Kornfeld D, Herd JA, et al: Biobehavioral variables and mortality or cardiac arrest in the Cardiac Arrhythmia Pilot Study (CAPS). Am J Cardiol 1990; 66:59–62Crossref, Medline, Google Scholar
3. Shapiro PA, Lesperance F, Frasure-Smith N, O’Connor CM, Baker B, Jiang JW, Dorian P, Harrison W, Glassman AH: An open-label preliminary trial of sertraline for treatment of major depression after acute myocardial infarction (the SADHART Trial). Am Heart J 1999; 137:1100–1106Crossref, Medline, Google Scholar
4. Glue P: SSRI and sympathomimetic interaction (letter). Br J Psychiatry 1996; 168:653; correction: 169:116Crossref, Medline, Google Scholar
5. Montgomery SA: New developments in the treatment of depression. J Clin Psychiatry 1999; 60(suppl 14):10–15; discussion 31–35Google Scholar
6. Serebruany VL, Gurbel PA, O’Connor CM: Platelet inhibition by sertraline and N–desmethylsertraline: a possible missing link between depression, coronary events, and mortality benefits of selective serotonin reuptake inhibitors. Pharmacol Res 2001; 43:453–462Crossref, Medline, Google Scholar
7. Neuger J, Wistedt B, Sinner B, Aberg-Wistedt A, Stain-Malmgren R: The effect of citalopram treatment on platelet serotonin function in panic disorders. Int Clin Psychopharmacol 2000; 15:83–91Crossref, Medline, Google Scholar
8. Serebruany VL, O’Connor CM, Gurbel PA: Effect of selective serotonin reuptake inhibitors on platelets in patients with coronary artery disease. Am J Cardiol 2001; 87:1398–1400Crossref, Medline, Google Scholar
9. Koe BK: Preclinical pharmacology of sertraline: a potent and specific inhibitor of serotonin reuptake. J Clin Psychiatry 1990; 51(suppl B):13–17Google Scholar
10. Glassman AH, O’Connor CM, Califf RM, Swedberg K, Schwartz P, Bigger JT Jr, Krishnan KR, van Zyl LT, Swenson JR, Finkel MS, Landau C, Shapiro PA, Pepine CJ, Mardekian J, Harrison WM, Barton D, McIvor M (SADHEART Group): Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709; correction: 288:1720Crossref, Medline, Google Scholar
11. Serebruany VL, Glassman AH, Malinin AI, Nemeroff CB, Musselman DL, van Zyl LT, Finkel MS, Krishnan KR, Gaffney M, Harrison W, Califf RM, O’Connor CM (SADHART Study Group): Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events. Circulation 2003; 108:939–944; correction: 108:3165Crossref, Medline, Google Scholar
12. Serebruany VL, Glassman AH, Malinin AI, Atar D, Sane DC, Oshrine BR, Ferguson JJ, O’Connor CM: Selective serotonin reuptake inhibitors yield additional antiplatelet protection in patients with congestive heart failure treated with antecedent aspirin. Eur J Heart Fail 2003; 5:517–521Crossref, Medline, Google Scholar
13. Suckow RF, Zhang MF, Cooper TB: Sensitive and selective liquid-chromatographic assay of fluoxetine and norfluoxetine in plasma with fluorescence detection after precolumn derivatization. Clin Chem 1992; 38:1756–1761Medline, Google Scholar
14. Serebruany VL, Gurbel PA, Shustov AR, Ohman EM, Topol EJ: Heterogeneity of platelet aggregation and major surface receptor expression in patients with acute myocardial infarction. Am Heart J 1998; 136:398–405Crossref, Medline, Google Scholar
15. Serebruany VL, Gurbel PA, Shustov AR, Dalesandro MR, Gumbs CI, Grabletz LB, Bahr RD, Ohman EM, Topol EJ: Depressed platelet status in an elderly patient with hemorrhagic stroke after thrombolysis for acute myocardial infarction. Stroke 1998; 29:235–238Crossref, Medline, Google Scholar
16. Gurbel PA, Serebruany VL, Shustov AR, Bahr RD, Carpo C, Ohman EM, Topol EJ (GUSTO-III Investigators): Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. J Am Coll Cardiol 1998; 31:1466–1473Crossref, Medline, Google Scholar
17. Briley MS, Raisman R, Sechter D, Zarifian E, Langer SZ: [3H]-Imipramine binding in human platelets: a new biochemical parameter in depression. Neuropharmacology 1980; 19:1209–1210Crossref, Medline, Google Scholar
18. Hrdina PD, Bakish D, Ravindran A, Chudzik J, Cavazzoni P, Lapierre YD: Platelet serotonergic indices in major depression: up-regulation of 5-HT2A receptors unchanged by antidepressant treatment. Psychiatry Res 1997; 66:73–85Crossref, Medline, Google Scholar
19. Dwivedi Y, Janicak PG, Pandey GN: Elevated [3H]inositol 1,4,5-trisphosphate binding sites and expressed inositol 1,4,5-trisphosphate receptor protein level in platelets of depressed patients. Psychopharmacology (Berl) 1998; 138:47–54Crossref, Medline, Google Scholar
20. Schneider LS, Severson JA, Pollock V, Cowan RP, Sloane RB: Platelet monoamine oxidase activity in elderly depressed outpatients. Biol Psychiatry 1986; 21:1360–1364Crossref, Medline, Google Scholar
21. Wahlund B, Saaf J, Wetterberg L: Classification of patients with affective disorders using platelet monoamine oxidase activity, serum melatonin and post-dexamethasone cortisol. Acta Psychiatr Scand 1995; 91:313–321Crossref, Medline, Google Scholar
22. Reichborn-Kjennerud T, Lingjaerde O, Oreland L: Platelet monoamine oxidase activity in patients with winter seasonal affective disorder. Psychiatry Res 1996; 62:273–280Crossref, Medline, Google Scholar
23. Butler J, Leonard BE: The platelet serotonergic system in depression and following sertraline treatment. Int Clin Psychopharmacol 1988; 3:343–347Crossref, Medline, Google Scholar
24. Alderman CP, Moritz CK, Ben-Tovim DI: Abnormal platelet aggregation associated with fluoxetine therapy. Ann Pharmacother 1992; 26:1517–1519Crossref, Medline, Google Scholar
25. Pai VB, Kelly MW: Bruising associated with the use of fluoxetine. Ann Pharmacother 1996; 30:786–788Crossref, Medline, Google Scholar
26. Skop BP, Brown TM: Potential vascular and bleeding complications of treatment with selective serotonin reuptake inhibitors. Psychosomatics 1996; 37:12–16Crossref, Medline, Google Scholar
27. Willerson JT, Eidt JF, McNatt J, Yao SK, Golino P, Anderson HV, Buja LM: Role of thromboxane and serotonin as mediators in the development of spontaneous alterations in coronary blood flow and neointimal proliferation in canine models with chronic coronary artery stenoses and endothelial injury. J Am Coll Cardiol 1991; 17(6 suppl B):101B-110BGoogle Scholar
28. Alderman CP, Seshadri P, Ben-Tovim DI: Effects of serotonin reuptake inhibitors on hemostasis. Ann Pharmacother 1996; 30:1232–1234Crossref, Medline, Google Scholar
29. Berk M, Jacobson BF, Hurly E: Fluoxetine and hemostatic function: a pilot study. J Clin Psychiatry 1995; 56:14–16Medline, Google Scholar
30. Nelva A, Guy C, Tardy-Poncet B, Beyens MN, Ratrema M, Benedetti C, Ollagnier M: [Hemorrhagic syndromes related to selective serotonin reuptake inhibitor (SSRI) antidepressants: seven case reports and review of the literature]. Rev Med Interne 2000; 21:152–160 (French)Crossref, Medline, Google Scholar
31. Bourdeaux R, Desor D, Lehr PR, Younos C, Capolaghi B: Effects of fluoxetine and norfluoxetine on 5-hydroxytryptamine metabolism in blood platelets and brain after administration to rats. J Pharm Pharmacol 1998; 50:1387–1392Crossref, Medline, Google Scholar

