
Does Donepezil Treatment Slow the Progression of Hippocampal Atrophy in Patients With Alzheimer’s Disease?
Abstract
OBJECTIVE: The only approved pharmacological approach for the symptomatic treatment of Alzheimer’s disease in Japan is the use of a cholinesterase inhibitor, donepezil hydrochloride. Recent in vivo and in vitro studies raise the possibility that cholinesterase inhibitors can slow the progression of Alzheimer’s disease. The purpose of the present study was to determine whether donepezil has a neuroprotective effect in Alzheimer’s disease by using the rate of hippocampal atrophy as a surrogate marker of disease progression. METHOD: In a prospective cohort study, 54 patients with Alzheimer’s disease who received donepezil treatment and 93 control patients with Alzheimer’s disease who never received anti-Alzheimer drugs underwent magnetic resonance imaging (MRI) twice at a 1-year interval. The annual rate of hippocampal atrophy of each subject was determined by using an MRI-based volumetric technique. Background characteristics, age, sex, disease duration, education, MRI interval, apolipoprotein E (APOE) genotype, and baseline Alzheimer’s Disease Assessment Scale score were comparable between the treated and control groups. RESULTS: The mean annual rate of hippocampal volume loss among the treated patients (mean=3.82%, SD=2.84%) was significantly smaller than that among the control patients (mean=5.04%, SD=2.54%). Upon analysis of covariance, where those confounding variables (age, sex, disease duration, education, MRI interval, APOE genotype, and baseline Alzheimer’s Disease Assessment Scale score) were entered into the model as covariates, the effect of donepezil treatment on hippocampal atrophy remained significant. CONCLUSIONS: Donepezil treatment slows the progression of hippocampal atrophy, suggesting a neuroprotective effect of donepezil in Alzheimer’s disease.
At present, the only approved pharmacological approach for the symptomatic treatment of Alzheimer’s disease in Japan is the use of cholinesterase inhibitors. Donepezil hydrochloride has been demonstrated to have significant effects in slowing symptomatic progression in 24-week placebo-controlled trials (1), and some long-term studies have shown that there is no less benefit after 1 year of treatment (2, 3). One observational study (4) showed that cholinesterase inhibitor treatment alters the natural history of Alzheimer’s disease, as indicated by the delay in admission to nursing homes. Furthermore, a longitudinal neuroimaging study using single photon emission computed tomography (SPECT) demonstrated that treatment of patients with Alzheimer’s disease with donepezil for 1 year reduced the decline in regional cerebral blood flow (rCBF) (5), suggesting the preservation of functional brain activity in donepezil-treated patients. Although these studies appear to have demonstrated the efficacy of donepezil in slowing down clinical disease progression, it is not clear whether donepezil treatment slows disease progression in Alzheimer’s disease.
Neuropathologically, Alzheimer’s disease is characterized by the presence of neurofibrillary tangles and senile plaques, impaired synaptic function, and cell loss (6). Although these histological features cannot be examined noninvasively, the cell loss that accompanies them can be seen in vivo as atrophy with magnetic resonance imaging (MRI). Among the characteristic neuropathological changes in Alzheimer’s disease, the most prominent structural changes at the initial stage occur in the hippocampal formation (7, 8). MRI-based volumetry of the medial temporal lobe structures has been proposed as a useful tool for the clinical diagnosis of Alzheimer’s disease (8). Serial MRI studies permit calculation of rates of atrophy over time. It has been proposed that measurement of the rate of atrophy could be used to monitor the effectiveness of antidementia drugs for Alzheimer’s disease (9, 10). If an anti-Alzheimer drug can slow down the anatomic progression of Alzheimer’s disease pathology, this should be detectable as a decrease in the rate of hippocampal atrophy in treated patients.
The purpose of the present study was to determine whether a cholinesterase inhibitor, donepezil hydrochloride, has a neuroprotective effect on Alzheimer’s disease. Using an MRI-based volumetric technique, we examined the rates of hippocampal atrophy in donepezil-treated Alzheimer’s disease patients and compared the results with those in control patients.
Method
Subjects and Study Design
In the present study, a prospective cohort was compared with a historical control cohort. All procedures followed the clinical study guidelines of the ethics committee of Hyogo Institute for Aging Brain and Cognitive Disorders, a research-oriented hospital, and were approved by the institutional review board. Written informed consent was obtained from the patients or their families.
The control group was selected among those who participated in the annual follow-up program before donepezil was introduced. Patients with Alzheimer’s disease who were examined at the Hyogo Institute for Aging Brain and Cognitive Disorders were invited to the Hyogo Institute’s Alzheimer’s disease annual follow-up program starting in July 1993. The consent and availability of a reliable caregiver and the absence of severe behavioral problems impelling institutionalization were the requirements for participating in the program. At baseline, the patients were examined comprehensively by both neurologists and psychiatrists under a short-term admission to the infirmary and were given routine laboratory tests, EEGs, and standardized neuropsychological examinations. The patients’ medical history was systematically collected from reliable family members by using a database format. Other imaging studies, such as MRIs of the brain, magnetic resonance angiograms of the head and neck, and cerebral perfusion or metabolism studies with positron emission tomography or SPECT were performed at baseline. Neuropsychological tests and MRIs were repeated at 1-year intervals. The clinical and investigative data collected prospectively in a standardized fashion were all added to the database (11).
After donepezil hydrochloride was licensed and marketed in November 1999 in Japan, the annual follow-up program was revised to the donepezil follow-up program because most patients with Alzheimer’s disease were treated with donepezil. The treated group consisted of consecutive patients with mild to moderate Alzheimer’s disease who received donepezil treatment and were enrolled in a prospective longitudinal cohort study between November 1999 and June 2003. Treatment with donepezil was a prerequisite for participating in the revised program. After the same baseline assessments as the annual follow-up program were evaluated, the patients received 3 mg/day of donepezil for 1 or 2 weeks and then 5 mg/day (the approved maximum dose in Japan). Donepezil hydrochloride was prescribed for the entire year after the initial MRI, and compliance was monitored at every visit (every 3 months). Neuropsychological tests and MRIs were repeated at 1-year intervals. The treated patients satisfied six inclusion criteria:
| 1. | Meeting criteria of the National Institute of Neurological Disease and Stroke/Alzheimer’s Disease and Related Disorders Association for probable Alzheimer’s disease (12) | ||||
| 2. | Having minimal to moderate functional severity (a score of 15 or more on the Mini-Mental State Examination (MMSE) (13) | ||||
| 3. | Having no lifetime history of other neurological or mental disorders | ||||
| 4. | Taking no established and documented antidementia drugs other than donepezil (agents with possible antidementia properties, such as nonsteroidal antiinflammatory drugs, vitamins E, ginkgo biloba, and lecithin were allowed) | ||||
| 5. | Having no evidence of focal brain lesions on a MRI | ||||
| 6. | There being informed consent from patients and their relatives for determination of their apolipoprotein E (APOE) genotype | ||||
The control group was selected among those who participated in the annual follow-up program between July 1993 and October 1999. Requirements for inclusion in the present study were the same as those for the donepezil follow-up participants except for the use of donepezil. Those who received anti-Alzheimer drugs other than donepezil were not included in the present study. In this study, only baseline and 1-year follow-up data were used.
Cognitive Functions
The status of global cognitive function was assessed with the MMSE and the Alzheimer’s Disease Assessment Scale (14). The Alzheimer’s Disease Assessment Scale was administrated by neuropsychologists (along with the MRI examination) who were not involved in managing the patients at a 1-year interval (10–14 months).
MRI Volumetry
We directly measured the volume of the hippocampal formation on high spatial resolution three-dimensional spoiled-gradient echo images (15). The images were generated perpendicular to the anterior-posterior commissure plane that covers the whole calvaria with a 1.5-T MRI unit (Sign Advantage 5.x, General Electric Medical Systems, Milwaukee). The operating parameters were as follows: field of view=220 mm, matrix=256×256, 124×1.5 mm contiguous sections, TR=14 msec, TE=3 msec, and flip angle=20°.
The detailed hippocampal MRI volumetric procedure is described elsewhere (15). In brief, the MRI data set was transmitted to a computer from the MRI unit, and after an appropriate data conversion, it was analyzed by using the public domain Image version 1.62 program (developed at the National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) with residential macro programs developed in our institute. Hippocampal formation volume in all subjects was measured by a single investigator (M.H.) who was blinded to 1) all clinical information, 2) the order (initial and follow-up) of MRIs, and 3) the time of enrollment. Before starting the measurement, the rater inspected the suitability of MRIs for hippocampal volumetry. If either one of the two MRIs obtained for a subject was unsuitable for volumetry because of motion artifacts or for any other technical reason, the subject was excluded from the study.
For volumetry, we used a combination of semiautomatic segmentation-through-density thresholding and manual tracing, thereby lowering partial voluming and the observer’s bias. The reliability and validity of volumetry have been established and described elsewhere (9, 15). The hippocampal formation was defined to include the pes hippocampi, digitationes hippocampi, alveus hippocampi, dentate gyrus, and subiculum (16–18). The boundary of the hippocampal formation was defined from the entire head of the hippocampus to the slice where the crus of the fornix was seen in full profile. The outline of the hippocampal formation together with the surrounding white matter and CSF was first traced with a manually guided mouse cursor, and subsequently, the gray matter of each structure was extracted by density thresholding set at a range between minimum and maximum pixel values. The maximum value was defined as the largest value for any pixel of the gray matter (represented by the caudate head). The minimum value was defined as one-half the sum of the mean pixel value of the gray matter and the mean value of CSF (represented by the lateral ventricle) (16). The slice volume of the hippocampal formation was obtained by automatically counting the number of pixels within the segmented regions and then multiplying the number by the voxel size: 9([220/256]2 × [1.5=1.1078 mm3]). The average volume of the right and left hippocampal formations was used for the analysis. The test-retest reliability (kappa=0.98) was measured as an intraclass correlation coefficient derived from three repeated measurements by a single rater under blinded conditions in 10 randomly selected subjects. Coefficients of variation (standard deviation/mean, where standard deviation indicates square root value of the arithmetic mean of 10 variance estimates) were 2.84% and 2.78% for the right and left hippocampal formations, respectively.
Determination of APOE Genotype
The detailed method for APOE genotyping is described elsewhere (19). In brief, genomic DNA was extracted from peripheral blood with a Genomix DNA extraction kit (Talent Corp., Trieste, Italy) according to the manufacturer’s protocol. The APOE genotype was determined by using polymerase chain reaction restriction fragment length polymorphism, according to the procedure described by Wenham and colleagues (20).
Statistical Analysis
The annual rate of hippocampal atrophy was defined as the percentage change, which was computed as baseline hippocampal formation volume minus 1-year hippocampal formation volume divided by baseline hippocampal formation volume (×100). The change in cognitive decline was expressed as the difference (points) between the baseline and 1-year Alzheimer’s Disease Assessment Scale scores. Because the APOE e4 allele is known to be specifically related to hippocampal atrophy in Alzheimer’s disease (9, 21), the effect of donepezil on hippocampal atrophy is possibly influenced by the APOE genotype. Therefore, we used two-way analysis of variance, which included the effects of donepezil treatment (i.e., treated and untreated), APOE type (i.e., presence and absence of the APOE e4 allele), and their interaction. When the interaction term was not significant, the effects of the donepezil treatment were examined with a t test. When significant effects were found, they were further examined with analysis of covariance (ANCOVA) in which age, sex, disease duration, education, interval between the two MRI studies (days), APOE genotype, and baseline MMSE score were entered into the model as covariates. The level of significance was set at p<0.05 for all statistical analyses.
Results
Twelve of 77 patients who were initially enrolled in the donepezil follow-up program were dropped from the study. Seven withdrew their consent, two were institutionalized, and the remaining three withdrew for other reasons. Of the 65 patients who completed the entire procedure of the study, 11 were excluded because the quality of one of their two MRIs was unsuitable for volumetry. Thus, 54 patients remained in the treated group. Two patients did not receive the full, 1-year dosage of donepezil. One patient discontinued donepezil after 3 weeks because of adverse effects, and the other took the drug irregularly (receiving about 60% of the full dose). However, these patients were included in the primary analyses.
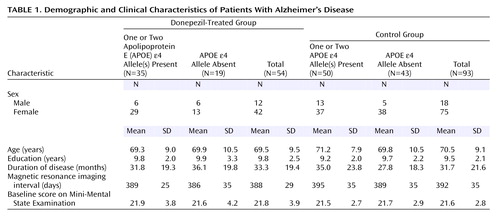
Of the 108 patients who completed the entire procedure of the annual follow-up study, 15 patients were excluded because their MRIs were of poor quality. Thus, 93 patients remained in the control group. The background characteristics of both groups are summarized in Table 1. The treated and control groups were not significantly different in age, sex, education, disease duration, intervals between the first and second MRI, or baseline MMSE scores.
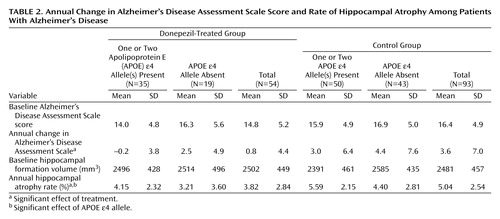
Hippocampal volumes and Alzheimer’s Disease Assessment Scale scores are summarized in Table 2. The effect of donepezil treatment on the change in Alzheimer’s Disease Assessment Scale scores was significant (F=5.55, df=143, p<0.02), but neither the effect of the APOE e4 allele (F=3.64, df=143, p<0.06) nor the interaction term (F=0.32, df=143, p=0.57) was significant. The effects of donepezil treatment and the APOE e4 allele on the rate of hippocampal atrophy were significant (F=8.19, df=143, p=0.005, and F=5.29, df=143, p<0.03, respectively), but the interaction term was not significant (F=0.08, df=143, p=0.78). In a further analysis, the mean annual rate of hippocampal atrophy in the treated group (mean=3.82%, SD=2.84%) was significantly lower than in the control group (mean=5.04%, SD=2.54%) (t=–2.7, df=145, p=0.008). In a one-way ANCOVA, where age, sex, disease duration, education, MRI interval, APOE genotype, and baseline Alzheimer’s Disease Assessment Scale score were entered into the model as covariates, the effect of donepezil on hippocampal atrophy remained significant (F=10.34, df=138, p=0.002).
Even if the two patients who did not receive the full 1-year dosage of donepezil were excluded from the analyses, the results remained unchanged; donepezil treatment had significant effects on Alzheimer’s Disease Assessment Scale score (F=4.37, df=141, p<0.04) and the rate of hippocampal atrophy (F=6.53, df=141, p<0.02), and the APOE e4 allele had a significant effect on the rate of hippocampal atrophy (F=4.14, df=142, p<0.05). The mean annual rate of hippocampal atrophy in the treated group (mean=3.93%, SD=2.76%) was significantly lower than in the control group (mean=5.04%, SD=2.54%) (t=–2.4, df=143, p<0.02).
Discussion
In the present study, the mean annual decline in the Alzheimer’s Disease Assessment Scale score in the control group (3.6 points) was significantly larger than in the treated group (0.8 points). Thus, these results are consistent with the results of previous long-term studies of donepezil (2, 3), and suggest that treatment with donepezil for 1 year was associated with a cognitive benefit. Moreover, donepezil seemingly slows the rate of hippocampal atrophy. The decrease in hippocampal volume in the treated patients (3.82%) was about 24% less than in the control patients (5.04%). The present results suggest that donepezil has both symptomatic and disease-progression slowing effects. The mean annual rate of hippocampal atrophy was significantly higher in the patients with the APOE e4 allele than in those without the APOE e4 allele, replicating our previous observation that the APOE e4 allele is specifically related to accelerated hippocampal pathology in Alzheimer’s disease (9). On the other hand, for the rate of hippocampal atrophy and for the change in Alzheimer’s Disease Assessment Scale, no significant interaction was noted between donepezil treatment and the APOE genotype. In a previous study, the effect of donepezil treatment did not appear to be affected by the APOE genotype (22). Our data are consistent with this observation.
In recent studies, long-term donepezil treatment has been demonstrated to slow the decline in cognition and functional activities (2, 3), delay the admission to a nursing home (4), and reduce the decline in rCBF (5). However, these studies do not answer the question of whether the beneficial effects of donepezil are due to symptomatic suppression, which leaves the underlying disease process unaltered, or to neuroprotection modifying the disease process. Recently, one randomized, double-blind, placebo-controlled pilot study in patients with Alzheimer’s disease demonstrated that donepezil-treated patients had significantly smaller mean decreases in total hippocampal volumes compared with placebo-treated patients. Although that study had limitations in that it included a relatively small number of patients and the period of drug treatment was short, our data are consistent with the results (23). Furthermore, two clinical trials, both open-label, placebo-controlled extension studies, have raised the possibility that cholinesterase inhibitors have slowing effects on both symptomatic and disease progression in Alzheimer’s disease patients. In one study (24), the level of cognitive function in patients who were treated with placebo for 3 months, followed by 12 months of donepezil treatment, was lower than in patients who were treated with donepezil for all 15 months. In the other study (25), the level of cognitive function of the patients who were treated with placebo for 26 weeks, followed by 26 weeks of rivastigmine treatment, was lower than that in patients who were treated with rivastigmine for all 52 weeks. The inability to catch up in those in whom the treatment with cholinesterase inhibitors was postponed reminds us of a potential disease-modifying effect.
A recent long-term randomized, double-blind trial (26) showed that no significant benefits were seen with donepezil compared with placebo in the institutionalization and progression of disability. In the study, there were no significant differences in behavioral symptoms, caregiver well-being, and caregiver time costs. On the other hand, statistically significant cognitive and functional effects were maintained over at least 2 years. Because many of the outcomes are influenced by the interaction of complex biological, social, and environmental factors, they might be inappropriate for assessing the neuroprotective effects of donepezil.
To explain the neuroprotective effect of cholinesterase inhibitors, mechanisms based on beta-amyloid metabolism have been postulated. Accumulation of amyloid is one of the earliest changes in Alzheimer’s disease pathology (27, 28) and may cause neuronal death in the CNS (29, 30). In vitro studies have demonstrated a link between cholinergic activation and beta-amyloid precursor protein metabolism. Wallace et al. (31) found evidence that lesions of the cholinergic nucleus basalis of Meynert increased the synthesis of beta-amyloid precursor protein in the cerebral cortex of rats. Wolf et al. (32), using human CNS neurons, found increased amyloid precursor protein secretion and decreased beta-amyloid protein production with carbachol stimulation of muscarinic receptors (32). These studies support a beneficial alteration in amyloid processing associated with cholinergic stimulation. Kihara et al. (33) examined the effects of nicotinic receptor agonists on amyloid beta cytotoxicity in cultured rat cortical neurons and found that nicotinic receptor stimulation may be able to protect neurons from degeneration induced by amyloid beta. Svensson and Nordberg (34) demonstrated that tacrine and donepezil at clinically relevant concentrations attenuated amyloid beta (25-35)-induced toxicity in rat pheochromocytoma PC12 cells. The neuroprotective effect was blocked by the presence of the nicotinic antagonists mecamylamine and tubocurarine, suggesting an intervention through nicotinic receptors.
Our study has some limitations. First, because it was a comparative study with a historical control group rather than a randomized study, it suffers from several sources of bias. The groups are not comparable because of the selection of subjects who received the intervention (selection bias), the cointerventions and other medical management being received by the two groups were different (performance bias), and the methods of outcome measurement being used in the two groups were different (detection bias). Although there was no intention to select subjects for the control and treatment groups, patients who could not tolerate the initial doses of donepezil were not included in the donepezil follow-up program. This could have been a source of selection bias. However, we are unaware of any evidence that donepezil tolerance predicts disease progression. Because the control group in the present study was not concurrent but historical, there was a generation difference of several years between the groups. Although the generation difference was a possible source of selection and performance bias, the difference in the patients’ generation was not likely to affect the volumetric results, and the cointerventions and other medical management did not change throughout the first and second halves of the study period. Second, it has been recognized that open-label studies are not optimal. The fact that the investigators were not blinded to treatment might increase the donepezil effect. However, neither hippocampal volume nor the Alzheimer’s Disease Assessment Scale score was a subjective outcome measure. Furthermore, in the present study, the Alzheimer’s Disease Assessment Scale was administered by neuropsychologists who were unblinded but not involved in managing the patients, and MRI volumetry was made by an investigator who was blinded to all clinical information. Therefore, it was unlikely that detection bias affected the results of the rate of hippocampal atrophy or the Alzheimer’s Disease Assessment Scale score in favor of the donepezil-treated group. Third, dropouts are a problem for this type of design and a possible source of exclusion bias. Simply ignoring everyone who has withdrawn from a clinical trial may bias the results, usually in favor of the intervention. Therefore, it is standard practice to analyze the results of comparative studies on an intention-to-treat basis. In the present study, two patients who did not take the full 1-year dosage of donepezil because of adverse events or poor compliance were included in the primary analyses.
As a result, the patients’ background characteristics, such as age, sex, education, disease duration, disease severity, and MRI interval, were comparable between the two groups, and even after we controlled for these variables, the effect of donepezil on hippocampal atrophy remained unchanged. In any case, the significant difference of the rate of hippocampal atrophy was likely to be due to the intervention with donepezil. Although our findings should be confirmed by a randomized, controlled long-term trial in patients with Alzheimer’s disease, it would be unethical to conduct such a study to extend the knowledge of donepezil.
The present results suggest that donepezil has not only a symptomatic effect but also a neuroprotective effect. If donepezil does, in fact, influence disease progression, we need to modify our treatment strategies; donepezil is not an optional but rather a mandatory treatment for Alzheimer’s disease and should be started in the prodromal or very early stage of the disease. Mild cognitive impairment is a condition that frequently progresses to Alzheimer’s disease, which requires early diagnostic and therapeutic interventions. Donepezil, through its neuroprotective effect, could possibly inhibit progression from mild cognitive impairment to Alzheimer’s disease. Further studies are needed to determine whether donepezil slows the progression from mild cognitive impairment to Alzheimer’s disease. Hippocampal atrophy could be used as a surrogate marker of disease progression in such studies. Furthermore, the potential neuroprotective mechanism should be refined and exploited to enhance the drug’s effectiveness in treating Alzheimer’s disease. A better understanding of this mechanism may suggest strategies for designing improved drugs.
 |
 |
Received June 11, 2004; revision received Sept. 1, 2004; accepted Sept. 10, 2004. From the Hyogo Institute for Aging Brain and Cognitive Disorders, Hyogo, Japan; Sawa Hospital at Toyonaka; the Department of Psychiatry and Behavior Science, Osaka University Graduate School of Medicine, Osaka, Japan; the Department of Psychiatry and Neurology, Kobe Graduate University School of Medicine, Hyogo, Japan; and the Department of Behavioral Neurology and Cognitive Neuroscience, Tohoku University Graduate School of Medicine, Sendai, Japan. Address correspondence and reprint requests to Dr. Hashimoto, Sawa Hospital, 1-9-1 Shiroyamacho, Toyonaka, Osaka, 561-8691, Japan; [email protected] (e-mail).
1. Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT: A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998; 50:136–145Crossref, Medline, Google Scholar
2. Winblad B, Engedal K, Soininen H, Verhey F, Waldemar G, Wimo A, Wetterholm AL, Zhang R, Haglund A, Subbiah P (Donepezil Nordic Study Group): A 1-year, randomized, placebo-controlled study of donepezil in patients with mild to moderate AD. Neurology 2001; 57:489–495Crossref, Medline, Google Scholar
3. Mohs RC, Doody RS, Morris JC, Ieni JR, Rogers SL, Perdomo CA, Pratt RD (“312” Study Group): A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology 2001; 57:481–488Crossref, Medline, Google Scholar
4. Lopez OL, Becker JT, Wisniewski S, Saxton J, Kaufer DI, DeKosky ST: Cholinesterase inhibitor treatment alters the natural history of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2002; 72:310–314Crossref, Medline, Google Scholar
5. Nakano S, Asada T, Matsuda H, Uno M, Takasaki M: Donepezil hydrochloride preserves regional cerebral blood flow in patients with Alzheimer’s disease. J Nucl Med 2001; 42:1441–1445Medline, Google Scholar
6. Braak H, Braak E: Pathology of Alzheimer’s disease, in Neurodegenerative Disease. Edited by Calne DB. Philadelphia, WB Saunders, 1994, pp 585–613Google Scholar
7. Laakso MP, Soininen H, Partanen K, Helkala EL, Hartikainen P, Vainio P, Hallikainen M, Hanninen T, Riekkinen PJ Sr: Volumes of hippocampus, amygdala and frontal lobes in the MRI-based diagnosis of early Alzheimer’s disease: correlation with memory functions. J Neural Transm 1995; 9:73–86Crossref, Medline, Google Scholar
8. Jack CR Jr, Petersen RC, O’Brien PC, Tangalos EG: MR based hippocampal volumetry in the diagnosis of Alzheimer’s disease. Neurology 1992; 42:183–188Crossref, Medline, Google Scholar
9. Mori E, Lee K, Yasuda M, Hashimoto M, Kazui H, Hirono N, Matsui M: Accelerated hippocampal atrophy in Alzheimer’s disease with apolipoprotein E e4 allele. Ann Neurol 2002; 51:209–214Crossref, Medline, Google Scholar
10. Jack CR Jr, Petersen RC, Xu Y, O’Brien PC, Smith GE, Ivnik RJ, Tangalos EG, Kokmen E: Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology 1998; 51:993–999Crossref, Medline, Google Scholar
11. Imamura T, Hirono N, Hashimoto M, Shimomura T, Tanimukai S, Kazui H, Hanihara T, Mori E: Clinical diagnosis of dementia with Lewy bodies in a Japanese dementia registry. Dement Geriatr Cogn Disord 1999; 10:210–216Crossref, Medline, Google Scholar
12. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM: Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34:939–944Crossref, Medline, Google Scholar
13. Folstein MF, Folstein SE, McHugh PR: “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198Crossref, Medline, Google Scholar
14. Mohs RC, Rosen WG, Davis KL: The Alzheimer’s Disease Assessment Scale: an instrument for assessing treatment efficacy. Psychopharmacol Bull 1983; 19:448–450Medline, Google Scholar
15. Mori E, Yoneda Y, Yamashita H, Hirono N, Ikeda M, Yamadori A: Medial temporal structures related to memory impairment in Alzheimer’s disease: an MRI volumetric study. J Neurol Neurosurg Psychiatry 1997; 63:214–221Crossref, Medline, Google Scholar
16. Lencz T, McCarthy G, Bronen RA, Scott TM, Inserni JA, Sass KJ, Novelly RA, Kim JH, Spencer DD: Quantitative magnetic resonance imaging in temporal lobe epilepsy: relationship to neuropathology and neuropsychological function. Ann Neurol 1992; 31:629–637Crossref, Medline, Google Scholar
17. Lehéricy S, Baulac M, Chiras J, Pierot L, Martin N, Pillon B, Deweer B, Dubois B, Marsault C: Amygdalohippocampal MR volume measurement in the early stages of Alzheimer disease. Am J Neuroradiol 1994; 15:927–937Google Scholar
18. Jack CR Jr: MRI-based hippocampal volume measurements in epilepsy. Epilepsia 1994; 35(suppl 6):21–29Google Scholar
19. Yasuda M, Maeda K, Shimada K, Kakigi T, Mori E, Nakai M, Nishio H, Tanaka C: Apolipoprotein E e4 allele and gender difference in risk of Alzheimer’s disease. Alzheimer Res 1995; 1:77–81Google Scholar
20. Wenham PR, Price WH, Blandell G: Apolipoprotein E genotyping by one-stage PCR. Lancet 1991; 337:1158–1159Crossref, Medline, Google Scholar
21. Hashimoto M, Yasuda M, Tanimukai S, Matsui M, Hirono N, Kazui H, Mori E: Apolipoprotein E e4 and the pattern of regional brain atrophy in Alzheimer’s disease. Neurology 2001; 57:1461–1466Crossref, Medline, Google Scholar
22. Greenberg SM, Tennis MK, Brown LB, Gomez-Isla T, Hayden DL, Schoenfeld DA, Walsh KL, Corwin C, Daffner KR, Friedman P, Meadows ME, Sperling RA, Growdon JH: Donepezil therapy in clinical practice: a randomized crossover study. Arch Neurol 2000; 57:94–99Crossref, Medline, Google Scholar
23. Krishnan KR, Charles HC, Doraiswamy PM, Mintzer J, Weisler R, Yu X, Perdomo C, Ieni JR, Rogers S: Randomized, placebo-controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer’s disease. Am J Psychiatry 2003; 160:2003–2011Link, Google Scholar
24. Doody RS, Geldmacher DS, Gordon B, Perdomo CA, Pratt RD (Donepezil Study Group): Open-label, multicenter, phase 3 extension study of the safety and efficacy of donepezil in patients with Alzheimer disease. Arch Neurol 2001; 58:427–433Crossref, Medline, Google Scholar
25. Farlow M, Anand R, Messina J Jr, Hartman R, Veach J: A 52-week study of the efficacy of rivastigmine in patients with mild to moderately severe Alzheimer’s disease. Eur Neurol 2000; 44:236–241Crossref, Medline, Google Scholar
26. Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, Edwards S, Hardyman W, Raftery J, Crome P, Lendon C, Shaw H, Bentham P (AD2000 Collaborative Group): Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomized double-blind trial. Lancet 2004; 363:2105–2115Crossref, Medline, Google Scholar
27. Hardy JA, Allsop D: Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 1991; 12:383–388Crossref, Medline, Google Scholar
28. Hardy JA, Higgins GA: Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256:184–185Crossref, Medline, Google Scholar
29. Yankner BA, Duffy LK, Kirschner DA: Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990; 250:279–282Crossref, Medline, Google Scholar
30. Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW: Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA 1993; 90:7951–7955Crossref, Medline, Google Scholar
31. Wallace W, Ahlers ST, Gotlib J, Bragin V, Sugar J, Gluck R, Shea PA, Davis KL, Haroutunian V: Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation. Proc Natl Acad Sci USA 1993; 90:8712–8716Crossref, Medline, Google Scholar
32. Wolf BA, Wertkin AM, Jolly YC, Yasuda RP, Wolfe BB, Konrad RJ, Manning D, Ravi S, Williamson JR, Lee VM: Muscarinic regulation of Alzheimer’s disease amyloid precursor protein secretion and amyloid beta-protein production in human neuronal NT2N cells. J Biol Chem 1995; 270:4916–4922Crossref, Medline, Google Scholar
33. Kihara T, Shimohara S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A: Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol 1997; 42:159–163Crossref, Medline, Google Scholar
34. Svensson AL, Nordberg A: Tacrine and donepezil attenuate the neurotoxic effect of A beta(25–35) in rat PC12 cells. Neuroreport 1998; 9:1519–1522Crossref, Medline, Google Scholar

