
Safety Reporting in Randomized Trials of Mental Health Interventions
Abstract
OBJECTIVE: The authors aimed to evaluate the adequacy of the reporting of safety information in publications of randomized trials of mental-health-related interventions. METHOD: The authors randomly selected 200 entries from the PsiTri registry of mental-health-related controlled trials. This yielded 142 randomized trials that were analyzed for adequacy and relative emphasis of their content on safety issues. They examined drug trials as well as trials of other types of interventions. RESULTS: Across the 142 eligible trials, 103 involved drugs. Twenty-five of the 142 trials had at least 100 randomly chosen subjects and at least 50 subjects in a study arm. Among drug trials, only 21.4% had adequate reporting of clinical adverse events, and only 16.5% had adequate reporting of laboratory-determined toxicity, while 32.0% reported both the numbers and the reasons for withdrawals due to toxicity in each arm. On average, drug trials devoted 1/10 of a page in their results sections to safety, and 58.3% devoted more space to the names and affiliations of authors than to safety. None of the trials of nondrug interventions had adequate or even partially adequate reporting of either clinical adverse events or laboratory-determined toxicity. In multivariate modeling, long-term trials and trials conducted in the United States devoted even less space to safety, while schizophrenia trials devoted more space to safety than did trials in other areas. CONCLUSIONS: Safety reporting is largely neglected across trials of mental-health-related interventions, thus hindering the assessment of risk-benefit ratios for rational decision making in mental health care.
Determining adverse events and their frequency for various pharmacological and other interventions is often a challenge (1–4). Randomized trials offer a prime opportunity for collecting not only efficacy data but also useful information on adverse events (1–3, 5, 6). However, empirical evaluations have shown that the reporting of safety information, including withdrawals due to toxicity, clinical adverse events, and laboratory-determined toxicity, is often neglected in randomized controlled trials of therapeutic and preventive interventions (7–11). These deficiencies have serious consequences, limiting the ability to understand the risk-benefit ratios of these interventions (12), even when they prove to be effective. Previous work on safety reporting has addressed trials in various medical fields (7–11). There is evidence that some deficiencies are more prominent in trials from some medical areas than in others (7). Nevertheless, to our knowledge, none of the previous evaluations has specifically targeted trials of interventions in the area of mental health. In this work, we have evaluated safety reporting in a large random sample of randomized controlled trials on various mental-health-related interventions.
Method
Trial Database
We used a random sample of trials derived from the PsiTri registry. PsiTri is a comprehensive registry of controlled trials across the field of mental health (13). It has been developed as a concerted action funded by the European Commission Quality of Life Programme, and it combines the specialist registries of five mental-health-related collaborative review groups of the Cochrane Collaboration that are working on depression, anxiety, and neurosis; developmental, psychosocial, and learning problems; dementia and cognitive improvement; schizophrenia; and abuse of drugs and alcohol. The PsiTri registry is based on trials rather than references; i.e., references of different reports that pertain to the same controlled trial are grouped under the same trial entry. In December 2002, when sampling was performed, the registry contained 16,504 controlled trials. The registry is evolving, with the continuous addition of more trials, but it is estimated that over 90% of trials in the field are already included. We randomly selected 200 controlled trial entries from the registry. We stratified the random selection by collaborative review group (corresponding to the respective mental health diagnoses) to ensure that the proportion of diseases in the selected sample is representative of the overall registry. We then excluded trials that were controlled but not randomized and trials without peer-reviewed journal publications because it is impossible to access the coverage of safety information simply from abstracts. We included all trials, regardless of sample size. Whenever there were several peer-reviewed publications on the same trial, we decided which was the main trial publication (the one presenting the most complete version of data on the primary outcomes of the trial) and examined whether the other reports offered any additional information on safety parameters. Both the main report and any additional published information were considered in the appraisal of the adequacy of safety reporting. Whenever a single report presented two or more trials with distinct, independent randomizations, we considered each trial separately.
Parameters of Safety Reporting
We used both qualitative and quantitative components of adverse event reporting that have been previously validated (7, 8). Qualitative components include 1) whether the number of withdrawals and discontinuations of study treatment due to toxicity is reported, 2) whether the number is given for each specific type of side effect leading to withdrawal, and 3) whether the severity of the described clinical adverse events and abnormalities of laboratory tests (laboratory-determined toxicity) is adequately defined, only partially defined, or inadequately defined. Adequate definition requires either detailed description of the severity or reference to a known scale of toxicity severity (typically with grades defined as 1=mild, 2=moderate, 3=severe, 4=life-threatening), with at least separate reporting of severe or higher-grade events; at least two adverse events (clinical or laboratory) have to be defined in this way, with numbers or rates given per study arm. Partial definition means that the reported numbers or rates mixed grade 2 with higher toxicity counts or the number of severe or higher grades of toxicity cases are separately specified only for one of many reported adverse events and laboratory abnormalities. Inadequate definition includes protocols lumping numbers of various types of adverse events; those lumping numbers for all grades of toxicity without separating any grades for any specific adverse events; those providing only generic statements (e.g., “the medication was not well tolerated”); and those not reporting anything regarding harm.
Quantitative measures assess the relative emphasis given to safety in the results of published trials (7). These measures are 1) the extent of space (absolute numbers of printed pages and proportion) devoted to safety in the results sections; 2) pages devoted to safety compared with pages devoted to the names and affiliations of authors, participants, and contributors in the same trial report; and 3) the use of tables and/or figures containing safety information. The space for each section was measured with a 0.05-page resolution (7).
Data were extracted by one investigator (P.N.P.) and discussed with a second investigator (J.P.A.I.) whenever any information was deemed unclear. For the assessment of the adequacy of reporting of clinical adverse effects and laboratory-determined toxicity, the kappa coefficients between independent data extractors have been previously estimated to be 0.72 and 0.85, respectively (8). In a sample of 60 trials, there were no instances in which one extractor considered the reporting adequate and the other inadequate. Moreover, we observed no important discrepancies in data extraction of quantitative parameters.
Other Trial Parameters
Trials were categorized first according to whether drugs were involved in the randomized comparisons or not. Nondrug interventions (e.g., psychological, behavioral, and social) may need different approaches to safety. We also separated trials that had at least 100 randomized subjects and over 50 subjects in a study arm. This group has been specifically targeted in previous evaluations of safety reporting (7, 8). Furthermore, for each eligible trial, we noted whether the trial showed significant differences (p<0.05) for any of the main efficacy endpoints considered, whether different doses/schedules of the same therapy were compared, and whether the study design compared only different doses/schedules; the exact trial sample size; masking (double blind or not); the population (predominantly adult versus pediatric); the mean duration of follow-up (long-term being more than 1 year); the main financial sponsor of the trial (government versus other/unreported); the location where most patients were recruited (United States versus other); and the year of publication.
Analyses
Analyses included descriptive assessments of the qualitative and quantitative parameters of safety reporting. Furthermore, regression analyses were conducted to assess parameters influencing safety reporting.
Descriptive data are reported for trials in which drugs were involved; for all studies; for trials with a sample size of at least 100 patients and at least 50 patients allocated to a study arm; and for trials published in 1990 or later.
All of the variables mentioned were used as predictors in least-squares regression models by using the percentage of space devoted to safety reporting in the results section as the dependent variable. Other regression analyses using logistic regression addressed the effect of these parameters on adequate reporting of clinical adverse events, laboratory-defined toxicity, and reasons for discontinuations due to toxicity per study arm. Multivariate models started from all variables with p<0.25 in univariate analyses and used backward elimination procedures. Only drug trials were included in the regression analyses. Regression analyses also examined whether any of the reporting parameters improved over time.
Statistical analyses were performed in SPSS (SPSS, Inc., Chicago). P values were two-tailed.
Results
Characteristics of Eligible Trials
Of 200 randomly selected Psi-Tri trial entries, we excluded 22 in which none of the references had been published in journals, 44 that did not pertain to randomized trials, and two that could not be retrieved in full text. Thus, 132 reports with 142 separate randomized trials (N=11,939 subjects) were eligible for further analysis. Of those, 11 had more than one journal publication (N=48 secondary publications). Only two secondary publications provided additional safety data, on one trial each. Trials belonged to the depression, anxiety, and neurosis (N=79); developmental, psychosocial, and learning problems (N=5); dementia and cognitive improvement (N=13); schizophrenia (N=33); and drug and alcohol (N=12) domains, respectively. Of the 142 trials, 25 had at least 100 subjects and at least 50 subjects in a study arm. Drugs were involved in the randomized comparisons in 103 (72.5%) of the 142 trials (N=7,963 subjects). Of the 103 drug trials, 62 contained an arm that received only placebo or no treatment. Nondrug trials addressed other biological (N=6), behavioral (N=13), social or health services (N=11), and psychological (N=9) interventions.
Overall, studies were of small group size (Table 1). About two-thirds of the trials were double blind, the percentages were higher among drug trials, and about a quarter involved some dose comparisons. Most trials showed statistically significant results for efficacy. About a third of the trials had received government funding. Few trials had long-term follow-up, and few involved children. Approximately half had been conducted in the United States. Trial reports covered a wide range of publication years, with half being published after 1990.
Qualitative Assessment of Safety Reporting
Overall, less than one out of six trials had adequate reporting of clinical adverse effects and even fewer trials had adequate reporting of laboratory-determined toxicity (Table 2). Studies with at least 100 subjects and at least 50 subjects in an arm had somewhat worse documentation of clinical adverse events and somewhat better documentation of laboratory-determined toxicity. Even among drug trials, the rates of adequate reporting were very low (21.4% and 16.5%, respectively). Drug trials with placebo/no treatment arms also had very low rates of adequate reporting (17.7% and 12.9%, respectively). No nondrug trials reported adequately on clinical adverse events or laboratory-determined toxicity.
Discontinuations due to toxicity per study arm were mentioned in slightly less than half of the trials, and the percentage was not much better in trials with at least 100 subjects or drug trials. Specific reasons for these discontinuations per arm were given less frequently, and this was recorded in only 8% of the trials with at least 100 subjects and at least 50 subjects in an arm. Nine nondrug trials simply stated clearly that no subjects discontinued the trial or mentioned reasons for discontinuations that were unrelated to toxicity. Otherwise, none of the nondrug trials provided data on discontinuations occurring due to toxicity.
There was no statistically significant heterogeneity in the rates of adequate reporting across collaborative review group domains for clinical adverse events (p=0.09), laboratory-determined toxicity (p=0.28), or the two withdrawal outcomes (p=0.72 and p=0.52) (all by Fisher’s exact test).
Quantitative Parameters of Safety Reporting
Little space was devoted to safety (Table 3). Even when nondrug trials were excluded, this was on average about 1/10 of a page or even less for trials with a placebo/no treatment arm (median=0.05 page). Large trials with at least 100 subjects and at least 50 subjects in an arm used only slightly more space (median=0.15 page). Overall, the space given to safety information was less than the space given for the names of authors and their affiliations (p<0.001). The difference remained significant when analyses were limited to drug trials, while for trials with at least 100 subjects and at least 50 subjects in an arm, the space for safety was similar to the space devoted to authors and affiliations. Tables and/or figures for safety were used infrequently, and even for exclusively drug trials.
Regression Analyses
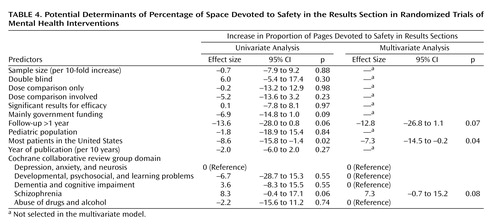
The percentage of space in the results sections dedicated to safety was significantly and independently larger in trials of schizophrenia, and it was independently smaller in trials conducted in the United States and in trials with long-term follow-up (Table 4). Adequate reporting for clinical adverse events was more common in trials of pediatric populations and less common in trials conducted in the United States (multivariate odds ratio=8.6, 95% confidence interval [CI]=1.2–62.0, p<0.04, and multivariate odds ratio=0.3, 95% CI=0.1–0.9, p<0.04, respectively). Adequate reporting of laboratory-determined toxicity was more common in larger trials (odds ratio=3.7, 95% CI=1.1–12.5 per 10-fold increase in sample size, p<0.04), but it was not significantly affected by any other parameters. Conversely, reporting of the reasons for discontinuations due to toxicity per study arm was less common in larger trials, and it was also independently less common in studies where dose comparisons were involved (multivariate odds ratio=0.2, 95% CI=0.1–0.6, p=0.004, and multivariate odds ratio=0.3, 95% CI=0.1–0.9, p<0.04, respectively).
The quality of safety reporting did not improve in recent trials (Table 2 and Table 3). In logistic regression, the odds of adequate reporting of clinical adverse events, laboratory-determined toxicity, and reasons for withdrawals due to toxicity nonsignificantly changed by 0.78-fold (p=0.29), 0.86-fold (p=0.54), and 0.94-fold (p=0.76) per 10 years, respectively, while the pages devoted to safety nonsignificantly decreased by 0.05 per 10 years (p=0.29).
Discussion
Randomized trials in the field of mental health often neglect the safety aspects of the tested interventions. Even with lenient criteria, very few drug trials and practically none of the nondrug trials have adequate reporting of clinical adverse events and laboratory-determined toxicity. Trial reports devote less space to safety than to names of authors and affiliations. Most trials in this field are of relatively small sample size (14) and may be underpowered to address safety issues, but even larger trials do not score consistently higher on all parameters of safety reporting. Very few trials in the field have long-term follow-up, and these seem to pay even less attention to safety relative to efficacy. If anything, less space has been given to safety in trials conducted in the United States, and safety reporting is not improving in more recent trials. We could not identify any parameters that have consistently led to substantially improving the reporting of both clinical and laboratory-determined toxicity as well as withdrawals.
The neglect of safety in mental health-related trials is similar and possibly worse than what has been reported for six different medical fields (HIV therapy, hypertension in the elderly, acute sinusitis, nonsteroidal anti-inflammatory drugs for rheumatoid arthritis, selective decontamination of the gastrointestinal tract, and antibiotics for Helicobacter pylori) (7). These previous evaluations had targeted exclusively trials with at least 100 subjects and at least 50 subjects in a randomized arm. Therefore, the most direct comparison should probably involve the 25 mental health trials fulfilling these sample size criteria. Even thus, the mental health field fared worse than all six other medical fields for the reporting of reasons for withdrawals and worse than five of six other medical fields for the reporting of clinical adverse events, while reporting rates for laboratory-determined toxicity were similar to the average of the six other fields. Withdrawals are frequent in mental health-related trials, and it is important to know why they have occurred (15).
In approximately one-quarter of the trials in our database, no drug treatments were evaluated. This constitutes a further difference between these trials and the traditional pharmacotherapy trials of many other medical domains. The consideration, let alone reporting, of adverse events in the assessment of nondrug interventions may be a challenge. Nondrug trials in our sample universally failed to report safety data. The adverse effects of behavioral, psychological, or social interventions are difficult to document and attribute to the tested intervention. However, attribution and causality is a problem even in drug trials (16). These obstacles should not prohibit the collection of data on adverse events. It is likely that psychosocial interventions can cause adverse outcomes. Unless investigators are prepared to record adverse events, these will never become known, and potentially harmful interventions may thus become established into mental health clinical practice.
In contrast to other medical areas (7), we found no improvement in the coverage of safety issues in trials involving dose comparisons. This is of concern since it suggests that dose-comparison trials in mental health place their emphasis unilaterally on efficacy. However, safety is an important consideration in selecting the dose or treatment schedule with the best benefit-to-harm ratio. Furthermore, the neglect of safety in long-term trials is also problematic, since long-term trials provide a key opportunity for understanding the safety of an intervention in a controlled setting with systematic recording of information. Finally, while it is encouraging that schizophrenia trials provided more attention to safety, this was true primarily for the amount of space and not for the quality of the information. Adverse events may be relatively more important in other mental diseases, which are less devastating than the major psychoses.
The neglect of safety reporting in randomized trials has implications for the conduct of evidence-based health care. Data suggest that not only clinical trials but also systematic reviews place little attention on toxicity (17, 18). Despite the fact that patients are usually poorly informed or unaware of even major adverse effects of the drugs that they are taking (19), patients would ideally wish to know a lot about potential toxicity, no matter how rare, before making therapeutic or preventive choices (20).
Some potential limitations should be discussed. We selected a random sample of trials from a registry that does not yet contain all mental-health-related trials. However, the registry coverage at the time of sampling probably exceeded 90%. Second, our sample included mostly small studies. Investigators may feel that small studies cannot make a meaningful contribution toward estimating the risk of uncommon or even common toxicities. However, the conduct of small studies is no justification for neglecting safety. For many interventions, only small trials will ever be performed. Third, it is difficult to reach a consensus on what constitutes an adequate reporting of safety. Space alone does not guarantee that important information is conveyed properly. We used standardized definitions that have been previously validated in other fields and that allowed comparisons with other medical areas. Qualitative and quantitative measures should provide complementary insights. Still, we encourage the further development of evaluation tools that would focus on appraising safety reporting in specific categories of trials, besides more generic tools (21). Nondrug trials in mental health are one area where such alternative tools would be particularly useful to develop. Fourth, some information on safety outcomes may be impossible to disentangle from efficacy outcomes. Mental health outcomes are often the integral composite of benefit and toxicity. Nevertheless, serious and life-threatening toxicity should be possible to record separately in most situations.
Finally, it is impossible to decipher whether the lack of safety information in a published article reflects the fact that such information was never collected or was collected but not reported. Given the fact that safety data are hard to retrieve after the trials are published (22), we strongly recommend that standardized reporting should be adopted (6, 23), with appropriate attention to toxicity. An extension of the CONSORT statement (23) for harm is currently being prepared. Information to contributors’ pages in mental health journals should emphasize the importance of covering information of potential harm in adequate detail with emphasis on serious and severe events and withdrawals per study arm.
 |
 |
 |
 |
Received July 31, 2003; revision received Nov. 19, 2003; accepted Nov. 21, 2003. From the Clinical Trials and Evidence-Based Medicine Unit, Department of Hygiene and Epidemiology, University of Ioannina School of Medicine; the Health Services Research Department, Institute of Psychiatry, London; the Department of Psychiatry, Lapinlahti Hospital, University of Helsinki, Helsinki, Finland; STAKES National Research and Development Centre for Welfare and Health, Helsinki; and the Institute for Clinical Research and Health Policy Studies, Department of Medicine, Tufts–New England Medical Center, Tufts University School of Medicine, Boston. Address reprint requests to Dr. Ioannidis, Department of Hygiene and Epidemiology, University of Ioannina School of Medicine, Ioannina 45110, Greece; [email protected] (e-mail). Supported by a concerted action grant from the European Commission Quality of Life Programme. The following participated in the EU-PSI Project: C. Adams, C. Almeida, J. Birks, S. Cardo, S. Gilbody, D. Hermans, M. Ferri, D.G. Contopoulos-Ioannidis, M. Davoli, J.A. Dennis, M. Ekqvist, M. Fenton, S. Leucht, G. MacDonald, H. McGuire, S. Pirkola, P. Pörtfors, R. Stancliffe, M. Starr, T.A. Trikalinos, A. Tuunainen, and S. Vecchi.
1. Enas GG, Goldstein DJ: Defining, monitoring and combining safety information in clinical trials. Stat Med 1995; 14:1099–1111Crossref, Medline, Google Scholar
2. Wallander MA: The way towards adverse event monitoring in clinical trials. Drug Saf 1993; 8:251–262Crossref, Medline, Google Scholar
3. Morse MA, Califf RM, Sugarman J: Monitoring and ensuring safety during clinical research. JAMA 2001; 285:1201–1205Crossref, Medline, Google Scholar
4. Edwards IR, Aronson JK: Adverse drug reactions: definitions, diagnosis, and management. Lancet 2000; 356:1255–1259Crossref, Medline, Google Scholar
5. O’Neill RT: Statistical analyses of adverse event data from clinical trials: special emphasis on serious events. Drug Inf J 1987; 21:9–20Crossref, Medline, Google Scholar
6. Ioannidis JP, Lau J: Improving safety reporting from randomised trials. Drug Saf 2002; 25:77–84Crossref, Medline, Google Scholar
7. Ioannidis JP, Lau J: Completeness of safety reporting in randomized trials: an evaluation of 7 medical areas. JAMA 2001; 285:437–443Crossref, Medline, Google Scholar
8. Ioannidis JPA, Contopoulos-Ioannidis DG: Reporting of safety data from randomised trials. Lancet 1998, 352:1752–1753Google Scholar
9. Loke YK, Derry S: Reporting of adverse drug reactions in randomised controlled trials—a systematic survey. BMC Clin Pharmacol 2001; 1:e3Google Scholar
10. Derry S, Loke YK, Aronson JK: Incomplete evidence: the inadequacy of databases in tracing published adverse drug reactions in clinical trials. BMC Med Res Methodol 2001; 1:e7Google Scholar
11. Edwards JE, McQuay HJ, Moore RA, Collins SL: Reporting of adverse effects in clinical trials should be improved: lessons from acute postoperative pain. J Pain Symptom Manage 1999, 18:427–437Google Scholar
12. Glasziou PP, Irwig LM: An evidence based approach to individualising treatment. Br Med J 1995; 311:1356–1359Crossref, Medline, Google Scholar
13. The EU-PSI Project, PsiTri, and the Mental Health Library. www.psitri.helsinki.fiGoogle Scholar
14. Thornley B, Adams C: Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ 1998; 317:1181–1184Crossref, Medline, Google Scholar
15. Wahlbeck K, Tuunainen A, Ahokas A, Leucht S: Dropout rates in randomised antipsychotic drug trials. Psychopharmacology (Berl) 2001; 155:230–233Crossref, Medline, Google Scholar
16. Naranjo CA, Busto U, Sellers EM: Difficulties in assessing adverse drug reactions in clinical trials. Prog Neuropsychopharmacol Biol Psychiatry 1982; 6:651–657Crossref, Medline, Google Scholar
17. Ernst E, Pittler MH: Systematic reviews neglect safety issues. Arch Intern Med 2001; 161:125–126Crossref, Medline, Google Scholar
18. Li Wan Po A, Herxheimer A, Poolsup N, Aziz Z: How do Cochrane reviewers address adverse effects of drug therapy? Cochrane Collaboration Methods Newsletter, June 2001, pp 20–21Google Scholar
19. Papanikolaou PN, Ioannidis JP: Awareness of the side effects of possessed medications in a community setting. Eur J Clin Pharmacol 2003; 58:821–827Crossref, Medline, Google Scholar
20. Ziegler DK, Mosier MC, Buenaver M, Okuyemi K: How much information about adverse effects of medications do patients want from physicians? Arch Intern Med 2001; 161:706–713Crossref, Medline, Google Scholar
21. Miller AB, Hoogstraten B, Staquet M, Winkler A: Reporting results of cancer treatment. Cancer 1981; 47:207–214Crossref, Medline, Google Scholar
22. Ioannidis JP, Chew P, Lau J: Standardized retrieval of side effects data for meta-analysis of safety outcomes: a feasibility study in acute sinusitis. J Clin Epidemiol 2002; 55:619–626Crossref, Medline, Google Scholar
23. Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne D, Gotzsche PC, Lang T: The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Ann Intern Med 2001; 134:663–694Crossref, Medline, Google Scholar

