
Linkage and Association of the Mitochondrial Aspartate/Glutamate Carrier SLC25A12 Gene With Autism
Abstract
OBJECTIVE: Autism/autistic disorder (MIM number 209850) is a complex, largely genetic psychiatric disorder. The authors recently mapped a susceptibility locus for autism to chromosome region 2q24-q33 (MIM number 606053). In the present study, genes across the 2q24-q33 interval were analyzed to identify an autism susceptibility gene in this region. METHOD: Mutation screening of positional candidate genes was performed in two stages. The first stage involved identifying, in unrelated subjects showing linkage to 2q24-q33, genetic variants in exons and flanking sequence within candidate genes and comparing the frequency of the variants between autistic and unrelated nonautistic subjects. Two single nucleotide polymorphisms (SNPs) that showed evidence for divergent distribution between autistic and nonautistic subjects were identified, both within SLC25A12, a gene encoding the mitochondrial aspartate/glutamate carrier (AGC1). In the second stage, the two SNPs in SLC25A12 were further genotyped in 411 autistic families, and linkage and association tests were carried out in the 197 informative families. RESULTS: Linkage and association were observed between autistic disorder and the two SNPs, rs2056202 and rs2292813, found in SLC25A12. Using either a single affected subject per family or all affected subjects, evidence for excess transmission was found by the Transmission Disequilibrium Test for rs2056202, rs2292813, and a two-locus G*G haplotype. Similar results were observed using TRANSMIT for the analyses. Evidence for linkage was supported by linkage analysis with the two SNPs, with a maximal multipoint nonparametric linkage score of 1.57 and a maximal multipoint heterogeneity lod score of 2.11. Genotype relative risk could be estimated to be between 2.4 and 4.8 for persons homozygous at these loci. CONCLUSIONS: A strong association of autism with SNPs within the SLC25A12 gene was demonstrated. Further studies are needed to confirm this association and to decipher any potential etiological role of AGC1 in autism.
Autism or autistic disorder (MIM number 209850) is a neurodevelopmental disorder characterized by a deficit in verbal and nonverbal communication, impairments in reciprocal social interactions, and patterns of repetitive or stereotyped behaviors and interests (1–3). The sex ratio is 4:1 male to female, and the prevalence of the disease is currently thought to possibly be above 1 per 1,000 persons (4). Autism appears to be the most highly genetic of the psychiatric disorders, as evidenced by the high risk of autism in additional children in families with an autistic child (estimated to be 50 to 100 times greater than that expected by chance) and the concordance rate for monozygotic twins being much higher than that of dizygotic twins (5). Heritability estimates of idiopathic autism are above 90% (6), so much of the disorder can be attributed to a genetic etiology. However, autism does not follow a simple Mendelian mode of transmission (i.e., dominant or recessive transmission) but is clearly a polygenic disease (4). A commonly accepted genetic model involves several genes (between five and 10) that interact to produce the disorder.
A genetic mutation or variant segregating with autism has yet to be unequivocally identified. Candidate genes for studies of autism range from genes that are thought to play a role in neurodevelopmental pathways, comportment, or behavior, such as genes in the serotonergic pathway or reelin (4, 7, 8). A few polymorphisms in several genes have been associated with the disorder in certain studies but not in others (4, 9–11).
Several independent studies involving genome-wide scans have now been published and point to significant linkage between autism and the chromosome 2q and 7q regions (4, 12). Our studies defined chromosome 2q24-q33 as a susceptibility region for autism with a peak at D2S335, which was particularly evident in families with more severe autism, as defined by delayed onset (>36 months) of phrase speech (MIM number 606053). We observed a nonparametric linkage score of 3.32, and a heterogeneity lod score (logarithm of the odds ratio for linkage) of 2.99 (13). Using a cohort of 152 autism sibling-pair families mostly from European countries, the International Molecular Genetic Study of Autism Consortium reported their highest multipoint lod score (3.74) at D2S2188 in families with autism with language delay (defined in that study as no single word before 24 months or no phrase speech before 33 months) (14). When stricter diagnostic criteria were used, the maximum lod score increased to a value of 4.8. Finally, a study from the Collaborative Autism Team using the phrase speech delay criterion to weight its data also showed a linkage between autism and chromosome region 2q33, with a maximum lod score of 2.86 and a heterogeneity lod score of 2.12 at D2S116 (15).
D2S335, D2S2188, and D2S116 are localized on chromosome 2 at 172.7 megabases (Mb), 175.8 Mb, and 201.8 Mb, respectively (Figure 1). This indicates that a critical region of susceptibility for autism occurs near D2S335 and D2S2188 in 2q31. In this interval, several known genes and expressed sequence tags have been mapped (Figure 1). Recently, the International Molecular Genetic Study of Autism Consortium has reported the analysis of nine candidate genes (TBR1, GAD1, DLX1, DLX2, cAMP-GEFII, CHN1, CREB2, HOXD1, and NEUROD1) localized across a 30-Mb region of 2q that are expressed in the CNS and encode proteins that play a role in neuronal cells or in neurobiological pathways (16). Variants were observed in TBR1, cAMP-GEFII, CHN1, HOXD1, and NEUROD1. However, no evidence was found that any of the candidate genes contributes to autism. In this region, our laboratory, together with the laboratory of Dr. Miriam Meisler, has previously investigated the neuronal voltage-gated sodium channels type I, II, and III (SCN1A, SCN2A, and SCN3A) in 117 multiplex autism families (17). Rare mutations were identified, each in single families, that were not observed in comparison subjects. These mutations, while of great interest, are not likely to account for the evidence of linkage observed in this region.
The aim of the present study was to identify an autism susceptibility gene in the 2q31 region. A systematic screen for genetic variants in affected individuals identified linkage and association between autism and single nucleotide polymorphisms (SNPs) in the SLC25A12 gene.
Method
Subjects
A total of 411 families were either recruited by The Seaver Autism Research Center at Mount Sinai (N=40), co-recruited by The Seaver Autism Research Center and the Autism Genetic Resource Exchange (18) (N=127), or recruited by the Autism Genetic Resource Exchange (N=244). All parents provided written informed consent, and potentially affected individuals were assessed with the Autism Diagnostic Interview–Revised (19). Individuals meeting Autism Diagnostic Interview–Revised criteria for autism (19) or borderline autism (13) were defined as affected. The cohort (18) and research diagnosis definitions used in the current study (13) have been previously described. The entire cohort of 411 families included blood samples from more than 2,000 individuals, including 720 affected individuals (671 with autism, 49 with borderline autism) and available parents and siblings. The 411 families consisted of 274 multiplex families (typically affected sibling pairs) and 137 trio families. DNA from blood samples or transformed cells were either isolated as detailed in Buxbaum et al. (13) or provided by the Autism Genetic Resource Exchange repository.
Mutation Screening
To investigate the involvement of positional candidate genes in autism, we performed a two-stage screen.
In the first stage, exonic and flanking DNA from 35 to 47 patients from 38 autistic families linked to the chromosome 2q24-q33 region were screened for genetic variants by single-strand conformation polymorphism and denaturing high-performance liquid chromatography. The rationale for this approach was to find variants in linked, affected individuals rather then rely on variants in public databases identified in unaffected individuals. Furthermore, the focus was on exonic sequence and intronic sequence adjacent to exons as being most likely to harbor functionally important variants.
Primers to amplify exons and flanking regions of positional candidate genes were designed using Primer3 software (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) and are available on request. The forward and reverse primers were chosen to produce amplicons of less than 500 base pairs. Larger exons, in particular the last exons of the genes, were divided over several amplicons, each with a size of less than 500 base pairs. Primers within flanking introns were situated 26 to 186 base pairs from the exons. A total of 82 exons of nine genes were analyzed, using 93 amplicons with an average size of approximately 310 nucleotides.
For single-strand conformation polymorphism, forward primers were labeled using 100 μCi [γP32]-ATP and 10 units of T4 polynucleotide kinase, according to the protocol provided by Invitrogen. Amplification was carried out using Ampli-Taq Gold Polymerase (Applied Biosystems, Foster City, Calif.) in a final volume of 10 μl, consisting of 10 ng of genomic DNA, 10 mM Tris-HCl, 50 mM KCl, 2.5 mM MgCl, 100 μM dNTPs, 10 μM radioactively labeled forward and unlabeled reverse primers, and 0.5 units of Taq Polymerase. Two μl of radioactively labeled polymerase chain reaction (PCR) product were then mixed with 2 μl of formamide blue loading buffer, denatured, and separated on a nondenaturing MDE 0.5X gel (according to the protocol of BioWhittaker). Single-strand conformation polymorphisms were detected by autoradiography.
For denaturing high-performance liquid chromatography, 8 μl of PCR product were screened on the WAVE Nucleic Acid Fragment Analysis System (according to the manufacturer’s protocols). Chromatographic parameters, appropriate analysis temperatures, and melting domains visualization were determined by WAVEMAKER software. Samples were analyzed using at least two mobile-phase temperatures to maximize the chances to identify polymorphisms.
Any amplicons in which variants were detected by either single-strand conformation polymorphism or denaturing high-performance liquid chromatography were then sequenced by direct fluorescent sequencing of purified PCR products using the BIG DYE dideoxy-terminator kit v3.1 (Applied Biosystems) and an ABI3100 DNA sequencer with Performance Optimized Polymer 6 (Applied Biosystems). Variants were then genotyped in 38 unrelated affected and in 100 nonaffected subjects. Allele frequencies were compared to identify variants with potentially altered frequencies (defined as p≤0.10) between ethnically matched affected and nonaffected subjects (to reduce the chances for false positives due to stratification).
In the second stage, variants with frequencies that were potentially altered between affected and nonaffected subjects were then analyzed in the entire cohort of more than 2,000 individuals from 411 autistic families. Genotyping was carried out by using the biplex DNA Invader method (Third Wave Technologies, Madison, Wisc.). The Invader assay is based on the hybridization of an oligonucleotide probe that completely matches a DNA target and the subsequent cleavage of the overlapping structure by Cleavase VIII, resulting in a target-specific product that is recognized by a fluorescence resonance energy transfer cassette (20). Specific oligonucleotide probes corresponding to wild type and mutated single nucleotide polymorphisms (SNPs) are associated with specific fluorophores, enabling simultaneous detection of both DNA sequences in a single well. For our studies, diluted aliquots of the PCR products were combined with Invader mix solution (Third Wave Technologies) and incubated for the cleavage reaction in a thermal cycler (PTC-100, MJ Research, Waltham, Mass.). The reaction product was then analyzed on a fluorescence plate reader (CytoFluor Series 4000, PerSeptive Biosystems, Framingham, Mass.) using the appropriate parameters of excitation and emission for each fluorophore.
Statistical Analysis
Two-tailed chi-square tests were used for comparisons of allelic frequencies and distributions of genotypes between autistic and nonautistic groups. Fischer’s exact test was performed when the number in a group was less than five. Hardy-Weinberg distribution was examined for each identified polymorphism in autistic and nonautistic groups.
Statistical analyses for Transmission Disequilibrium Tests were computed with TDT-GENEHUNTER (GENEHUNTER version 2.1, compiled to run on the Unix environment of Mac OS X) and the S-TDT (21) program. Two-locus Transmission Disequilibrium Test was carried out with TDT-GENEHUNTER using the TDT2 option, and the haplotypes were constructed by GENEHUNTER and verified manually to ensure structure. Haplotypes were determined on the basis of transmission patterns in families in which both parents were genotyped. Association analyses were also carried out using TRANSMIT v2.5.2 (22), which tests for association between genetic marker or haplotype and disease even when parental genotypes are unknown or haplotype phase is unknown. Association tests were carried out with TRANSMIT for all affected subjects and for one randomly selected affected subject, the latter studies being carried out with 500 replications.
For linkage analysis, we used GENEHUNTER PLUS (compiled to run on the Unix environment of Mac OS X). Heterogeneity lod scores and nonparametric linkage scores were calculated for both single and multipoint analyses. For heterogeneity lod scores, data were analyzed under both a dominant and recessive model, using 50% penetrance and a value of 0.001 for the disease allele frequency. Such an approach detects linkage under many different conditions irrespective of the “true” underlying inheritance pattern (23, 24).
Genotype relative risk was calculated for heterozygote and homozygote subjects as described (25).
Linkage disequilibrium was estimated using a D′ value calculated with the 2LD program (26).
Results
Genes across the 2q31 region were screened for association in two stages as detailed in the Method section. The genes analyzed included glutamate decarboxylase 1 (GAD1) (in collaboration with Drs. Shigeo Kure, Kiyoshi Kanno, and Yoichi Matsubara), four hypothetical proteins (FLJ13096, FLJ13984, PRO2037, and FLJ23462, recently identified as duodenal cytochrome b) (in collaboration with Drs. Paolo Gasparini and Massimo Carella), histone acetylase-1 (HAT-1) (in collaboration with Dr. Salah Uddin Qureshi), the cytoplasmic dynein subunit DNCI2, the aspartate/glutamate carrier SLC25A12, and the homeobox protein DLX2 (Figure 1). These candidate genes were chosen on the basis of their position relative to the positive linkage results from three studies (13–15), their expression in brain tissue, and, in some cases, their known function, their novelty, or the existence of related genes within the region of chromosome 7 showing linkage to autism. For this latter criterion, we note that the linked region of chromosome 7 contains genes paralogous to DNCI2, SLC25A12, DLX1, and DLX2 (i.e., DNCI1, SLC25A13, DLX5, and DLX6).
In the first stage, all known exons (with flanking intronic sequence) of these genes were screened by single-strand conformation polymorphism and denaturing high-performance liquid chromatography for variants in 35 to 47 unrelated individuals chosen from families showing linkage to D2S335 as described in the Method section. In the nine genes, 82 exons were screened and 29 SNPs were identified. Frequencies of each variant were then evaluated in autistic patients (using only one affected individual per family, N=38) and in 50 ethnically matched nonautistic subjects, after confirming that the distribution of allele frequencies were in Hardy-Weinberg equilibrium. Only two SNPs, both within the SLC25A12 gene, showed significant differences in allele frequencies between autistic and nonautistic subjects using both allele-based (p<0.004) and genotype-based (p<0.03) tests.
Within the SLC25A12 gene, we identified a total of five variants in the first stage screen (including the two meeting criteria for further study). Figure 1 presents the five variants of the SLC25A12 gene identified in 47 affected subjects linked to the chromosome 2q24-q33 region. The two polymorphisms meeting criteria in the first stage, rs2056202 (I3-21A/G) and rs2292813 (I16+70A/G), are G/A variants in flanking intronic sequence located 21 base pairs upstream of exon 4 and 70 base pairs downstream of exon 16, respectively. Two variants, a C-T variant at nucleotide 99 (rs1878583) and a G-A variant at nucleotide 1418, were within coding regions. G1418A changes arginine 473 to glutamine, while the C99T variant is silent. G1418A is a new SNP (i.e., not reported in the National Center for Biotechnology Information dbSNPs database) located in a region conserved across mammalian species, but the amino acid glutamine is observed in mice. The final variant appears in the 3′ untranslated region. We did not find in our sample SNP rs1059299, reported in the public database, which changes amino acid 600.
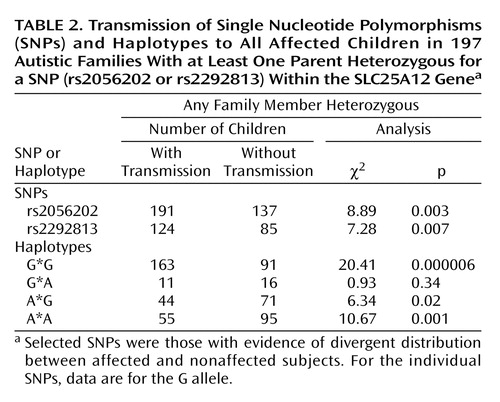
Given the evidence for association of rs2056202 and rs2292813 in a small number of affected and nonaffected subjects, the entire sample was genotyped at these SNPs for analysis by the Transmission Disequilibrium Test, which makes use of family-based comparison subjects. Of the 411 families studied, 197 had at least one parent heterozygous for at least one SNP. This group consisted of 140 multiplex and 57 singleton families. To test for association by the Transmission Disequilibrium Test, transmission from heterozygous parents to one affected child was analyzed (Table 1). Transmission Disequilibrium Test analysis demonstrated association for rs2056202 (p=0.001) and for rs2292813 (p=0.01). Similar excess transmission was assessed by TRANSMIT for rs2056202 (χ2=10.71, df=1, p=0.001) and rs2292813 (χ2=7.24, df=1, p=0.007). In both cases, the G allele appeared to be the risk allele (or the A allele the protective allele). For simplicity, Table 1 and Table 2 show transmission data for just the G allele for both SNPs.
Association studies were also carried out using multiple affected individuals per family (Table 2). Such analysis is more properly a measure of linkage rather than association, while providing increased power. Transmission disequilibrium was observed for both rs2056202 (p=0.003) and rs2292813 (p=0.007). Similar results were found with TRANSMIT for rs2056202 (χ2=10.3, df=1, p=0.001) and rs2292813 (χ2=8.17, df=1, p=0.004).
Looking at haplotypes, there was an increased transmission of the G*G haplotype in autism when analyzing either one affected individual per family (p=0.000003) (Table 1) or all affected individuals (p=0.000006) (Table 2). Using a global analysis, two-locus Transmission Disequilibrium Test showed disequilibrium of transmission of the four observed haplotypes for both one affected individual per family (χ2=32.31, df=3, p=0.0000005) or all affected individuals (χ2=28.76, df=3, p=0.000003). Similar observations were made with TRANSMIT for transmission of the G*G haplotype to either one affected subject (χ2=8.1, df=1, p=0.004) or all affected subjects (χ2=12.37, df=1, p=0.0004).
Genotype relative risk was estimated for individuals carrying one or two copies of the risk alleles (the G alleles for both SNPs). Using one affected subject per family, genotype relative risk could be estimated as 1.56 and 3 in heterozygotes and 2.51 and 4.81 in homozygotes for rs2056202 and rs2292813, respectively. Using all affected subjects, the values were 1.92 and 2 in heterozygotes and 2.36 and 2.88 in homozygotes. Note that estimates of genotype relative risk tend to be underestimated in family studies such as these (25).
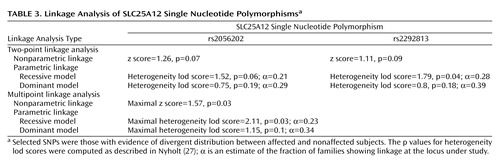
Two-point linkage analysis using nonparametric lod score analysis indicated some evidence for linkage between autism and rs2056202 or rs2292813 (Table 3). Two-point heterogeneity lod score supported this linkage, with maximal heterogeneity lod scores of 1.52 (p=0.06) and 1.79 (p=0.04) for rs2056202 and rs2292813, respectively. However, information was low at these SNPs (estimated as 0.21 and 0.28 for rs2056202 and rs2292813, respectively). To increase information we used multipoint linkage analyses with these two SNPs. Under these conditions, maximal multipoint nonparametric linkage scores of 1.57 and maximal multipoint heterogeneity lod scores of 2.11 were observed. The two markers showed linkage disequilibrium with each other as determined by analyzing linkage disequilibrium in unrelated patients (D′=0.79, SD=0.06).
To examine the relationship between the linkage and the association, we first identified a subset (selected from the 197 informative families) of 76 families that showed linkage, defined as a positive multipoint NPL value. In this subset, we found an increased transmission for rs2056202 with one affected per family (Transmission Disequilibrium Test: χ2=6.06, df=1, p=0.01; TRANSMIT: χ2=7.23, df=1, p=0.01) or all affected subjects (Transmission Disequilibrium Test: χ2=12.31, df=1, p=0.0005; TRANSMIT: χ2=14.64, df=1, p=0.0001) and for r2229813 with all affected subjects (Transmission Disequilibrium Test: χ2=8.91, df=1, p=0.003; TRANSMIT: χ2=9.24, df=1, p=0.002), but not with one affected subject (Transmission Disequilibrium Test: χ2=2.27, df=1, p=0.13; TRANSMIT: χ2=3.33, df=1, p=0.12). The G*G haplotype showed association with either one affected subject per family (Transmission Disequilibrium Test: χ2=7.14, df=1, p=0.008; TRANSMIT: χ2=8.97, df=1, p=0.004) or all affected subjects (Transmission Disequilibrium Test: χ2=21.25, df=1, p=0.000004; TRANSMIT: χ2=17.26, df=1, p=0.0006). In contrast, in the 121 informative families that did not show linkage, neither rs2056202 (for all affected subjects, Transmission Disequilibrium Test: χ2=1.28, df=1, p=0.26; TRANSMIT: χ2=2.03, df=1, p=0.15) nor rs2292813 (for all affected subjects, Transmission Disequilibrium Test: χ2=1.00, df=1, p=0.32; TRANSMIT: χ2=2.00, df=1, p=0.16) showed such evidence for association.
Discussion
The objective of the present work was to identify a susceptibility gene for autism in chromosome region 2q24-q33. We identified two SNPs, rs2056202 and rs2292813, in SLC25A12 that demonstrated association with autism using the Transmission Disequilibrium Test and TRANSMIT. Preferential transmission of the G allele for both SNPs was found in 197 informative families. Linkage was found between autism and the SNPs by Transmission Disequilibrium Test and by nonparametric and parametric analyses. The evidence for both linkage and association supports a role for SLC25A12 as an autism susceptibility gene (rather than a modifying gene). Furthermore, as the association was primarily evident in linked families, the data are consistent with there being a relationship between the observed linkage and the observed association.
The SLC25A12 gene contains 18 exons spread over about 110 kilobases (kb) and is expressed primarily as 2.9- and 3.2-kb mRNA species, predominantly in skeletal muscle, heart, and brain (28). SLC25A12 cDNA has an open reading frame of 2037 base pairs encoding a 678-amino acid protein that is a calcium-dependent mitochondrial aspartate/glutamate carrier (AGC1). The subcellular localization of protein is exclusively mitochondrial. The amino-terminal half of AGC1 contains five putative EF hands that are able to bind Ca2+, while the carboxy-terminal half harbors the aspartate/glutamate exchanger function. AGC1 is critically involved in the activity of the malate/aspartate NADH shuttle, catalyzing the electrogenic exchange of aspartate for glutamate and a proton, and is also involved in the urea cycle (29). Recently, it has been shown that AGC1 is the form of the mitochondrial aspartate/glutamate carrier expressed in neurons and neural stem cells (28). Protein levels increased during neuronal differentiation and are correlated with an increase in the malate/aspartate NADH shuttle activity.
SLC25A12 mRNA and the AGC1 protein are widely expressed in adult mouse CNS, particularly in neural nuclei in the brainstem (30). It has been suggested that the enrichments of AGC1 in specific neurons could reflect a tonic activity of these neurons. Dysfunction of this protein, or altered expression of this protein, may lead to an alteration in mitochondrial function and ATP synthesis. Since neurons are major energy users, even modest changes in mitochondrial function and ATP synthesis may lead to selective changes in neurons. Support for a role for mitochondrial dysfunction in autism comes from a recent study demonstrating mitochondrial hyperproliferation and partial respiratory chain block in two patients with autism and a 15q inverted duplication (31).
As noted earlier, a region of chromosome 7 has also been shown to be linked to autism in multiple studies (32). It is interesting that a paralog of SLC25A12, namely, SLC25A13 (CITRIN1 or AGC2), localizes to this region of chromosome 7. These two genes share about 79% identity, both encoding forms of aspartate/glutamate carriers, with 71% identity in the EF-hand domains and 84% within the exchanger domain. Mutations in the SLC25A13 gene confer adult-onset type II citrullinemia (CTLN2, MIM number 603471), an autosomal recessive disease caused by a deficiency of argino-succinate synthetase with clinical features that include enuresis diarrhea, tremors, lethargy, mental retardation, and psychiatric disorders. Although confirmation of the association between autism and SLC25A12 is required, it is tempting to speculate that SLC25A13 could be a candidate autism susceptibility gene on chromosome 7.
For both SNPs, the allele associated with autism corresponds to a common allele. One must consider that in complex disorders with multiple interacting genes, the prevalence of the susceptibility alleles may be quite high. Recent examples of this include the e4 allele of apoliprotein E in Alzheimer’s disease, NOD-2 gene changes in Crohn’s disease, and calpain-10 gene changes and type 2 diabetes mellitus (33–35). In the case of autism, with the strongest evidence for numerous interacting genes, the expectation would be that the susceptibility variants for at least some loci will be quite common and might even contribute to behavioral variability in healthy individuals. However, in any such study the true susceptibility locus or functional polymorphisms may not have been identified but rather are in linkage disequilibrium with the variants studied. The true susceptibility locus may even be in neighboring genes. In our studies, we have examined both flanking genes. For HAT1, we did not find any useful polymorphisms. For DNCI2, we found eight polymorphisms that were negative for association in first-stage analyses.
In our study, we identified intronic polymorphisms associated with disease. The functional relevance of such variants remains obscure in most studies. However, it is increasingly being realized that the expression of a significant number of genes is regulated by cis-acting elements and that inherited variation in gene expression may contribute to disease (36, 37). Regulation of gene expression would affect cellular function, without requiring a modification in the coding sequence, if levels of the gene product were limiting. It has recently been demonstrated that overexpression of AGC1 can lead to increased mitochondrial ATP production (38), so genetic variations that change the expression of AGC1 would be predicted to impact ATP production. Whether the polymorphisms we identified (or additional polymorphisms in linkage disequilibrium with the polymorphisms we identified) affect gene expression needs to be determined.
With all association studies, especially in complex disorders thought to be due to multiple interacting genes of weak effect, we must await replication in independent samples before the results can be accepted. However, given the evidence from our study, it may be unrealistic to expect that the finding will be easily replicated by Transmission Disequilibrium Test in a typical sample of under 200 trios. The Transmission Disequilibrium Test allows for robust statistical analysis without bias of population admixture. However, it lacks power, especially at loci such as ours, where many of the parents were homozygous at the two loci. A carefully designed case-control study, with control subjects matched for ethnicity, gender, and age, may have more power to detect association at these two loci. Alternatively, genotyping several hundred trios for Transmission Disequilibrium Test would be in order for a replication study.
Assuming that our results reflect true association of SLC25A12 with autism, the data indicate a genotype relative risk for a two-locus (G*G) haplotype of between 3 and 5. This, while significant, must be taken in context of the observation that the susceptibility variants (or the variants in linkage disequilibrium with the true susceptibility variants) are common alleles. This is consistent with the idea that this locus plays a significant role in the epidemiology of the disorder but would not be immediately useful for genetic counseling until it can be considered together with additional loci.
To further investigate SLC25A12 locus as a susceptibility gene for autism we are currently searching for additional genetic markers across the 110-kb region containing this gene, using both extant databases and sequencing in the patients we are studying, particularly of conserved noncoding regions. To date, more than 90 SNPs encompassing the SLC25A12 gene have been identified in the public databases, with at least 20 SNPs harboring a heterozygosity rate over 0.1. None of these appear in conserved noncoding regions. We are also carrying out expression studies of AGC1.
In summary, the current study presents evidence for association of SLC25A12 with autism. Further characterization of this gene and related genes, including SLC25A13, is in order.
 |
 |
 |
Received April 23, 2003; revision received Sept. 2, 2003; accepted Sept. 15, 2003. From the Laboratory of Molecular Neuropsychiatry, Departments of Psychiatry and Neurobiology, and The Seaver Autism Research Center, The Greater New York Autism Research Center of Excellence, Mount Sinai School of Medicine. Address reprint requests to Dr. Buxbaum, Department of Psychiatry, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1668, New York, NY 10029; [email protected] (e-mail). Supported by grants from the Seaver Autism Research Center and Cure Autism Now and by an NIMH Studies to Advance Autism Research and Treatment grant (MH-066673). The authors thank Cure Autism Now and the Autism Genetic Research Exchange for help and support; Dr. L. Alison McInnes for discussions and encouragement; Dr. David A. Greenberg for advice on data analysis; and the team of the Mount Sinai School of Medicine DNA Core Facility. The authors also thank Dr. Rudy Tanzi for providing help with ascertaining additional DNA samples. Last, the authors thank Joshua Lechner for implementing several critical programs on the OS X platform.

Figure 1. Genomic Organization of the Autism Susceptibility Locus in Chromosome Region 2q24-q33a
aFor the organization of positional candidate genes, arrows indicate orientation of transcription. Variants within the SLC25A12 gene identified in the present study are indicated (see text), with the two SNPs, rs2056202 (I3-21A/G) and rs2292813 (I16+70A/G), underlined.
1. Lord C, Leventhal BL, Cook EH Jr: Quantifying the phenotype in autism spectrum disorders. Am J Med Genet 2001; 105:36–38Crossref, Medline, Google Scholar
2. Lord C, Cook EH, Leventhal BL, Amaral DG: Autism spectrum disorders. Neuron 2000; 28:355–363Crossref, Medline, Google Scholar
3. Rapin I, Katzman R: Neurobiology of autism. Ann Neurol 1998; 43:7–14Crossref, Medline, Google Scholar
4. Folstein SE, Rosen-Sheidley B: Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet 2001; 2:943–955Crossref, Medline, Google Scholar
5. Rutter M: Concepts of autism: a review of research. J Child Psychol Psychiatry 1968; 9:1–25Crossref, Medline, Google Scholar
6. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M: Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995; 25:63–77Crossref, Medline, Google Scholar
7. Lucinio J, Alvarado J: Progress in the genetics of autism. Mol Psychiatry 2002; 7:1012–1017Crossref, Medline, Google Scholar
8. Philippe A, Guilloud-Bataille M, Martinez M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Penet C, Feingold J, Brice A, Leboyer M: Analysis of ten candidate genes in autism by association and linkage. Am J Med Genet 2002; 114:125–128Crossref, Medline, Google Scholar
9. Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH Jr, Fang F, Song C-Y, Vitale R: Association between a GABRB3 polymorphisms and autism. Mol Psychiatry 2002; 7:311–316Crossref, Medline, Google Scholar
10. Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T: Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry 2002; 7:302–310Crossref, Medline, Google Scholar
11. Kim SJ, Cox N, Courchesne R, Lord C, Corsello C, Akshoomoff N, Guter S, Leventhal BL, Courchesne E, Cook EH Jr: Transmission disequilibrium mapping at the serotonin transporter gene (SLC6A4) region in autistic disorder. Mol Psychiatry 2002; 7:278–288Crossref, Medline, Google Scholar
12. Veenstra-Vanderweele J, Cook E Jr, Lombroso PJ: Genetics of childhood disorders, XLVI: autism, part 5: genetics of autism. J Am Acad Child Adolesc Psychiatry 2003; 42:116–118Crossref, Medline, Google Scholar
13. Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL: Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet 2001; 68:1514–1520Crossref, Medline, Google Scholar
14. International Molecular Genetic Study of Autism Consortium (IMGSAC): A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet 2001; 69:570–581Crossref, Medline, Google Scholar
15. Shao Y, Raiford KL, Wolpert CM, Cope HA, Ravan SA, Ashley-Koch AA, Abramson RK, Wright HH, DeLong RG, Gilbert JR, Cuccaro ML, Pericak-Vance MA: Phenotypic homogeneity provides increased support for linkage on chromosome 2 in autistic disorder. Am J Hum Genet 2002; 70:1058–1061Crossref, Medline, Google Scholar
16. Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, Bailey AJ, Monaco AP, Maestrini E: Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol Psychiatry 2003; 8:916–924Crossref, Medline, Google Scholar
17. Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH: Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry 2003; 8:186–194Crossref, Medline, Google Scholar
18. Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ: The Autism Genetic Resource Exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet 2001; 69:463–466Crossref, Medline, Google Scholar
19. Lord C, Rutter M, Le Couteur A: Autism Diagnostic Interview—Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 1994; 24:659–685Crossref, Medline, Google Scholar
20. Olivier M, Chuang LM, Chang MS, Chen YT, Pei D, Ranade K, de Witte A, Allen J, Tran N, Curb D, Pratt R, Neefs H, de Arruda Indig M, Law S, Neri B, Wang L, Cox DR: High-throughput genotyping of single nucleotide polymorphisms using new biplex invader technology. Nucleic Acids Res 2002; 30:e53Google Scholar
21. Spielman RS, Ewens WJ: A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 1998; 62:450–458Crossref, Medline, Google Scholar
22. Clayton D: A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 1999; 65:1170–1177Crossref, Medline, Google Scholar
23. Durner M, Vieland VJ, Greenberg DA: Further evidence for the increased power of LOD scores compared with nonparametric methods. Am J Hum Genet 1999; 64:281–289Crossref, Medline, Google Scholar
24. Hodge SE, Vieland VJ, Greenberg DA: HLODs remain powerful tools for detection of linkage in the presence of genetic heterogeneity. Am J Hum Genet 2002; 70:556–559Crossref, Medline, Google Scholar
25. Risch N: Inplications of multilocus inheritance for gene-disease association studies. Theor Popul Biol 2001; 60:215–220Crossref, Medline, Google Scholar
26. Xie X, Ott J: Testing linkage disequilibrium between a disease gene and marker loci (abstract). Am J Hum Genet 1993; 53(suppl):110Google Scholar
27. Nyholt DR: All LODs are not created equal. Am J Hum Genet 200:282–288Google Scholar
28. del Arco A, Satrustegui J: Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem 1998; 273:23327–23334Crossref, Medline, Google Scholar
29. Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F: Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J 2001; 20:5060–5069Crossref, Medline, Google Scholar
30. Ramos M, del Arco A, Pardo B, Martinez-Serrano A, Martinez-Morales JR, Kobayashi K, Yasuda T, Bogonez E, Bovolenta P, Saheki T, Satrustegui J: Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res 2003; 143:33–46Crossref, Medline, Google Scholar
31. Filipek PA, Juranek J, Smith M, Mays LZ, Ramos ER, Bocian M, Masser-Frye D, Laulhere TM, Modahl C, Spence MA, Gargus JJ: Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol 2003; 53:801–804Crossref, Medline, Google Scholar
32. Badner JA, Gershon ED: Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Mol Psychiatry 2002; 7:56–66Crossref, Medline, Google Scholar
33. Botstein D, Risch N: Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 2003; 33(March suppl):228–237Google Scholar
34. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard J-P, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel J-F, Sahbatou M, Thomas G: Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001; 411:599–603Crossref, Medline, Google Scholar
35. Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Horikawa Y, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI: Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 2000; 26:163–175Crossref, Medline, Google Scholar
36. Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW: Allelic variation in human gene expression. Science 2002; 297:1143Crossref, Medline, Google Scholar
37. Bray NJ, Buckland PR, Owen MJ, O’Donovan MC: Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet 2003; 113:149–153Medline, Google Scholar
38. Lasorsa FM, Pinton P, Palmieri L, Fiermonte G, Rizzuto R, Palmieri F: Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J Biol Chem 2003; 278:38686–38692Crossref, Medline, Google Scholar

