
Clinical Features of Schizophrenia and Linkage to Chromosomes 5q, 6p, 8p, and 10p in the Irish Study of High-Density Schizophrenia Families
Abstract
OBJECTIVE: Schizophrenia is clinically heterogeneous. Recent linkage studies suggest that multiple genes are important in the etiology of schizophrenia. The authors examined the hypothesis of whether the clinical variability in schizophrenia is due to genetic heterogeneity. METHOD: Using data from the Irish Study of High-Density Schizophrenia Families (N=265 pedigrees; N=1,408 individuals), the authors attempted to predict, from major symptoms and signs of psychosis, evidence for linkage within families for schizophrenia-related disorders to chromosomal regions 5q21–5q31, 6p24–6p22, 8p22–8p21, and 10p15–10p11. RESULTS: No substantial evidence was found for associations between clinical features of schizophrenia and linkage to chromosomes 5q, 6p, or 10p. However, affected individuals from families with evidence for linkage to 8p had significantly more affective deterioration, poorer outcome, more thought disorder, and fewer depressive symptoms than affected individuals from the other families in the study. CONCLUSIONS: These results raise the possibility that the putative susceptibility gene for schizophrenia localized in the 8p22–8p21 region may predispose individuals to the core dementia-praecox syndrome described by Kraepelin more than 100 years ago.
Since the schizophrenic syndrome was first described as a diagnostic category (1, 2), clinicians have noted the extensive variability in its symptomatic presentation. A central question in schizophrenia research has therefore been whether clinical variability results from etiologic heterogeneity.
Evidence from twin, family, and adoption studies argues persuasively that genetic factors play a strong etiologic role in schizophrenia (3, 4). However, mathematical modeling of aggregate risk statistics and pedigree data usually fail to support the hypothesis that the genetic component to schizophrenia is due to a single genetic locus (4). Linkage studies of schizophrenia are now producing results consistent with these findings. That is, as an increasing number of genome scans for schizophrenia are being completed and published, no studies are finding strong evidence for a Mendelian-like locus (5–10). Rather, modest evidence for linkage is being found at a number of different genomic locations. Furthermore, in some studies, evidence for linkage heterogeneity has been found (9, 11). Thus, several lines of evidence suggest that several loci contribute to the vulnerability to schizophrenia.
A fundamental question to ask next is whether the clinical variability of schizophrenia arises from allelic variation at different susceptibility loci. Figure 1 illustrates two models that relate genetic and clinical heterogeneity for a complex disease such as schizophrenia, where the liability to illness is probably influenced by multiple genetic loci. In a common pathway model (figure 1, model A), all the genetic loci influence a common biological vulnerability, which in turn influences the occurrence of individual symptoms. This model might apply, for example, if all susceptibility genes were in a single biochemical or developmental pathway, and disruptions at any stage of the pathway would produce similar biological consequences. In an independent pathways model (figure 1, model B), the loci have direct and partially overlapping influences on specific disease symptoms. Thus, a susceptibility allele at locus 1 would produce high levels of symptoms 1 and 2, moderate levels of symptom 3, and no evidence of symptom 4, whereas a susceptibility allele at locus 3 would produce relatively high levels of symptoms 3 and 4, moderate levels of symptom 2, and no evidence of symptom 1. The independent pathways model might be correct if the distinct “mutations” that influenced risk for schizophrenia affected different neurobiological systems such as those that govern reality testing, perceptual distortion, and affective regulation.
Some evidence has accumulated that addresses the validity of these two hypotheses. In nearly all plausible genetic models for schizophrenia, affected relatives are highly likely to share the same susceptibility alleles. Under the independent pathways model, therefore, symptomatic resemblance should be seen in affected relative pairs. The first attempts to address this question, which examined whether Kraepelinian subtypes “ran true” in families, produced inconsistent results (12). Three more recent studies of this question, however, have all produced negative results (13–15). Efforts at examining whether specific clinical features of schizophrenia are correlated in pairs of affected relatives have been fewer but more successful (16–19). We recently examined resemblance for psychotic symptoms in affected sibling pairs from the Irish Study of High-Density Schizophrenia Families (20). Modest but significant sibling resemblance was seen for nearly all major psychotic symptoms; most correlations ranged from 0.10 to 0.30.
Although these more recent data are consistent with the independent pathways model, at least two other mechanisms could explain symptomatic resemblance in siblings with schizophrenia. First, they might be correlated for environmental variables that might influence symptoms such as perinatal complications, infectious agents, or access to treatment. Second, symptomatic resemblance may be due to other genes that do not directly influence the liability to schizophrenia, such as those affecting temperament or intelligence.
A more definitive way to evaluate the validity of these two models is to examine the association between individual symptoms and specific chromosomal regions thought to contain susceptibility loci. Two recent examples of this approach are noteworthy. In 10 Canadian high-density pedigrees, Brzustowicz et al. (21) found much stronger evidence of linkage to the proximal region of chromosome 6p by using a positive symptom scale as a quantitative measure than by examining the diagnosis of schizophrenia alone. More recently, Malhotra et al. (22) reported that the severity of hallucinations in drug-free patients with schizophrenia was significantly predicted by a functional polymorphism at the serotonin-transporter locus.
In this report, we take a different approach toward evaluating the common pathway versus the independent pathways models. We have, in the course of a genome scan in the Irish Study of High-Density Schizophrenia Families, uncovered evidence for susceptibility loci in four genomic regions: 5q21–5q31 (23), 6p24–6p22 (11), 8p22–8p21 (24), and 10p15–10p11 (25). Here we examine whether, in the 265 multiplex schizophrenia pedigrees from the high-density schizophrenia families study, there is an association between the evidence for linkage at these four loci and the level of major psychotic and affective symptoms displayed by the affected family members.
METHOD
Sample
As detailed elsewhere (26), the Irish Study of High-Density Schizophrenia Families is a collaborative project between the Medical College of Virginia, the Health Research Board, Dublin, and the Queen’s University, Belfast. Families containing a high density of schizophrenia and related disorders were ascertained through 39 separate public psychiatric hospitals in Ireland and Northern Ireland. No exclusion criteria were used in our selection of families. Pedigrees were not screened for bilineal descent of schizophrenia-spectrum illness or for the presence of other forms of psychopathology. However, compared with relatives of the epidemiologically sampled probands with schizophrenia from the Roscommon Family Study, relatives in the Irish Study of High-Density Schizophrenia Families had similar risks for affective illness, anxiety disorder, and alcoholism (26). The similar risk profile indicates that our ascertainment in the Irish Study of High-Density Schizophrenia Families did not select for families with high “loadings” for other major psychiatric disorders. Compared with epidemiologically ascertained probands with schizophrenia from a county in western Ireland (27), individuals with schizophrenia in the high-density families study had a similar symptom pattern except for greater levels of thought disorder, and fewer depressive symptoms; however, the high-density study individuals had a more chronic course and poorer outcome (26).
In this article, we report results from the linkage sample of the Irish Study of High-Density Schizophrenia Families (26), which includes all relatives for whom high-quality clinical information (personal interview and/or hospital record) and DNA were available (N=265 pedigrees; N=1,408 individuals). Genetic relationships were confirmed by using data from 25 microsatellite markers, a process that required reclassifying 13 full sibling pairs as half-siblings.
Procedure
Interviews were conducted by Irish psychiatrists and social scientists with a background in mental health or survey work after obtaining participants’ consent by using procedures approved by the ethical review panels at the Health Research Board and the Queens University. Whenever possible, individuals suspected of having psychosis were interviewed by a psychiatrist. All interviewers underwent extensive initial and ongoing training by the first author in the use of diagnostic instruments.
Our assessment instruments consisted of modified sections of the Structured Interview for DSM-III-R (28) for selected axis I disorders and the Structured Interview for Schizotypy (29) for putative schizophrenia-spectrum personality disorders. For 98.6% of the patients diagnosed with schizophrenia or schizoaffective disorder, we obtained psychiatric records. A detailed abstract of these records was dictated, and a specially developed case-record rating scale was completed. All relevant diagnostic information for each individual relative was reviewed independently by K.S.K. and D.W. These assessments were done blind to pedigree assignment and knowledge of the psychopathologic status of other relatives. Diagnostic disagreements were resolved by consensus. The agreement rate and weighted kappa (30) for our main system of 10 diagnostic categories in the high-density families study (26) were 73.3% and 0.94 (SD=0.05), respectively.
During the course of the blind diagnostic assessments, 11 key symptomatic and course variables from the Major Symptoms of Schizophrenia Scale (31) were assessed in all patients with nonaffective psychosis by one of these diagnosticians (K.S.K.) by using all available information. The key variables were: 1) hallucinations, 2) delusions (any), 3) Schneiderian delusions, 4) positive thought disorder (e.g., loosening of association), 5) catatonic symptoms, 6) affective deterioration (e.g., affective flattening), 7) negative thought disorder (e.g., alogia), 8) depressive symptoms, 9) manic symptoms, 10) chronicity of course (from single episode with recovery to chronic course without even partial remission), and 11) outcome (from full recovery to very poor outcome). These ratings reflected clinical judgment and incorporated the severity of the symptom, its duration, and its relative prominence over the entire course of the illness. Although outcome was coded on a 4-point scale, all other variables were coded on 5-point scales with the following guidelines: 1=clearly not present, 2=possibly present but subthreshold, 3=clearly present but moderate, 4=clearly present and prominent, and 5=clearly present and severe. Interrater reliability of the Major Symptoms of Schizophrenia Scale was tested on 47 cases of psychotic illness from the Roscommon Family Study rated blindly by K.S.K. and A. Gruenberg, M.D. Intraclass correlations ranged from 0.60 for catatonic symptoms to 0.91 for manic symptoms, with a mean of 0.77 (SD=0.11). In addition, we examined age at onset and gender.
In this report, we define psychotic illness as encompassing all cases of nonaffective psychosis (N=646), defined as individuals meeting DSM-III-R criteria for schizophrenia, schizoaffective disorder, schizophreniform disorder, delusional disorder, and psychosis not otherwise specified. In addition, we included in the category of nonaffective psychosis a small number of individuals that we diagnosed with simple schizophrenia by using criteria outlined elsewhere (31). Individuals meeting these criteria in the Roscommon Family Study appeared to suffer from a disorder that was related to schizophrenia, from both a clinical and familial perspective (31). Analyses were repeated examining only cases of DSM-III-R schizophrenia (N=542). A similar pattern of findings was obtained.
Linkage studies in the Irish Study of High-Density Schizophrenia Families have used three major categories of affection: 1) narrow=schizophrenia plus schizoaffective disorder, 2) intermediate=narrow plus schizotypal personality disorder and other nonaffective psychoses, and 3) broad=intermediate plus psychotic affective illness and other putative schizophrenia-spectrum personality disorders (26).
Statistical Analysis
We calculated, for each family, the multipoint lod score (logarithm of the odds ratio for linkage) over the relevant genomic region, using GENEHUNTER (32) under the genetic and phenotypic model that provided the strongest evidence for linkage in the entire sample. The genomic regions were defined by the following markers (centromeric to telomeric): 5q (D5S815–D5S658), 6p (D6S291–D6S477), 8p (D8S283–D8S552), and 10p (D10S183– D10S189). The phenotypic (and genetic) models used were as follows (11, 23–25): 5q—narrow (recessive), 6p—broad (additive), 8p—broad (dominant), and 10p—intermediate (recessive).
We examined two main approaches to the problem of associating evidence for linkage within a family to symptoms in affected individuals: utilizing the sign of the lod score (i.e., dividing families into those with positive versus negative evidence for linkage) or using the raw lod score itself. We evaluated these two approaches by predicting our symptom and course variables from both simultaneously, across each of the four chromosomal locations. The sign of the lod score was statistically significant in multiple analyses, but the raw lod score was never significant in the presence of the sign. These results suggested that the information about family “linkedness” was indexed by the sign of the family lod score rather that its precise quantitative estimate. Therefore, we divided our families into those with lod scores less than 0 and those with lod scores of 0 or more and compared the difference in individual symptoms between the affected members of these two groups of families by using a Wilcoxon test (33).
We considered the examination of two diagnostic classifications of affected individuals: those with schizophrenia and those with nonaffective psychosis. However, in regression analyses with family lod score as the dependent variable, including all relatives with nonaffective psychosis, a dummy independent variable reflecting whether that individual also met criteria for schizophrenia was never significant. These results indicated that all the predictive power was seen with the broader diagnostic category. We also explored eliminating families with lod scores between –0.10 and 0.10, dividing the families into three groups by lod scores, and using nonparametric indices of linkage instead of the lod score. Although the general pattern of results was similar, these other approaches did not give such marked levels of significance and are not reported here.
For age at onset, a comparison was made by using a t test. All p values were two-tailed. Although we performed a large number of analyses, they were highly correlated, making it difficult to implement a simple correction. In addition, we explicitly view these analyses as exploratory and in need of replication in other samples. Strictly speaking, the tests of significance assume a basis in a priori comparisons, which was not the case in the study reported here. As a compromise, we set the alpha level at 0.01.
To assess the magnitude of the association between individual clinical features and evidence for linkage, we standardized the symptom scores and age at onset so that differences between affected members of linked and unlinked families are expressed in standard deviation units.
RESULTS
Table 1 depicts the mean difference in standard deviation units in symptom score or age at onset for individuals affected with nonaffective psychosis in families with positive versus negative evidence for linkage to chromosomes 5q, 6p, 8p, and 10p. No significant results were obtained for linkage on 5q, 6p, or 10p. However, on chromosome 8p, affected individuals in the families with positive lod scores had higher levels of affective deterioration and thought disorder, worse outcome, and fewer depressive symptoms than affected individuals in families with negative lod scores. The effect sizes of these differences ranged from 0.25 to 0.34, meaning that on these four clinical features, affected individuals from families linked and not linked to the 8p region differed by between one-fourth and one-third of a standard deviation.
To explore further the relationship between symptom scores in affected individuals and the familial evidence for linkage to chromosome 8p, we divided the families into five groups by their lod score and examined mean scores for positive thought disorder, affective deterioration, depressive symptoms, and poor outcome in the affected individuals (figure 2). In no case was a clear monotonic function observed. For affective deterioration and poor outcome, and to some extent for positive thought disorder, the major difference was observed between members of families with positive versus negative lod scores.
DISCUSSION
The goal of this report was to use the clinical and linkage information from the Irish Study of High-Density Schizophrenia Families to evaluate two alternative hypotheses about the nature of the relationship between multiple putative susceptibility loci and clinical variability in schizophrenia. There are several methodological strengths of this investigation. First, the sample size of both families and affected individuals is large and ethnically homogeneous. Second, our clinical evaluations were based on both personal interview and hospital record data, increasing their likely accuracy. Third, we coded symptoms averaged over the clinical course of illness, thereby reducing the influence of fluctuations due to transient environmental factors. Fourth, the clinical evaluations were performed blind both to information about other family members and to evidence for linkage.
However, our study is also limited by several important methodological constraints. First, although other groups have reported linkages to each of the four regions studied (5q [34], 6p [35–37], 8p [9, 37], and 10p [38, 39]), in the absence of gene identification it is possible that one or more of these linkages represent false positives and do not contain susceptibility genes. Second, in the modest-sized families that predominate in the Irish Study of High-Density Schizophrenia Families, the lod score is a quite attenuated index of the true probability that a susceptibility allele is segregating in a given family. By chance alone, because of the small number of meioses available in a given family, a moderate proportion of unlinked families will appear linked and linked families will appear unlinked. Third, we have treated all affected individuals in linked families alike, assuming that they all carry the susceptibility allele. Given that environmental factors and other genetic loci likely also influence susceptibility to illness, this assumption is almost certainly incorrect. Some affected individuals from linked families will not share the major susceptibility gene with their affected relatives. Fourth, even though our symptom ratings were averaged over the disease course, there is little doubt that they are likely to be influenced by a range of factors over which we have no control, including environmental risk factors (e.g., perinatal complications and drug and alcohol abuse) and the quality of treatment. Fifth, our results might initially appear contradictory to our prior findings that linkage to the 8p region was maximized when a broad definition of the schizophrenia spectrum was used (24). The earlier results suggest that the susceptibility gene in the 8p region predisposes individuals to other psychotic conditions and schizophrenia-related personality disorders. However, the results reported here suggest that when psychotic illness is manifest, this gene appears to increase the probability that the illness will take a dementia-praecox-like course. Finally, we have performed a substantial number of analyses with these data. To avoid presenting many highly correlated tests, we selected both the form of linkage information (raw lod score versus the sign of the lod score) and diagnostic criteria (nonaffective psychosis versus schizophrenia) by using the quantitative criterion of relative significance level. However, these exploratory methods, which required examination of the relationship between symptoms and evidence for linkage, increase the risk for false-positive findings. It is noteworthy, therefore, that a significant relationship between affective deterioration, poor outcome, and linkage on 8p was observed regardless of which approach was taken toward diagnosis or linkage information.
Given these caveats, what can be concluded from these results? For chromosomes 5q, 6p, and 10p, no positive results emerged. By contrast, four significant findings were seen with the chromosome 8p linkage. These results deserve attention for two major reasons. First, several findings are at rather high levels of statistical significance (e.g., p<0.001), although these results are likely inflated because multiple tests were performed. Second, the results are clinically coherent. That is, families with positive evidence for linkage on 8p tend to contain affected individuals with clinical features that have long been associated with classical dementia praecox: thought disorder, affective deterioration, minimal depressive symptoms, chronic course, and poor outcome (40, 41). Furthermore, using latent class analysis we recently identified such a subtype of schizophrenia from a population-based sample of probands from the west of Ireland (42). However, the magnitude of the differences in these clinical features in affected individuals from linked versus unlinked families was modest and would be described as having small to medium effect sizes using Cohen’s definitions (43).
In conclusion, we have presented some evidence in favor of the independent pathways model for schizophrenia and against the common pathway model. In families from the Irish Study of High-Density Schizophrenia Families, evidence for linkage to chromosome 8p was significantly correlated with symptoms of classical dementia praecox in affected members. No such pattern was seen for linkage to the putative susceptibility regions on chromosomes 5q, 6p, and 10p. Our results suggest that individual genomic regions that may contain distinct susceptibility genes can be meaningfully related to the clinical variability of the schizophrenic syndrome. These results also provide additional limited evidence for the existence of a susceptibility gene in the 8p region, as otherwise, this pattern of findings would be difficult to explain.
However, the highly tentative nature of these interpretations is noteworthy. It is important that others attempt to confirm them in other high-density schizophrenia pedigree collections. In the future, we hope that we will be able to further examine the validity of these findings using high-risk marker haplotypes that would enable us to determine directly which individuals possess susceptibility alleles at the individual loci. Ultimate confirmation of these results must await discovery of the pathogenic alleles themselves.
Received Apr. 6, 1999; revision received Aug. 10, 1999; accepted Aug. 17, 1999From the Departments of Psychiatry and Human Genetics and the Virginia Institute for Psychiatric and Behavioral Genetics, Medical College of Virginia of Virginia Commonwealth University; the Department of Psychiatry, Mater Hospital, Belfast, Northern Ireland; and the Health Research Board, Dublin, Ireland. Address reprint requests to Dr. Kendler, Department of Psychiatry, Box 980710, Medical College of Virginia, Richmond, VA 23298-0710; [email protected] (e-mail). Supported by NIMH grants MH-41953 and MH-45390. Dr. Kendler was supported by a Research Scientist Award from NIMH (MH-01277). Data collection was conducted under the supervision of S. Humphries, M. Healy, and A. Finnerty. Additional interviews were conducted by John Burke, Fiona Duke, Rosemary Shinkwin, M. Ni Nuallain, F. McMahon, J. Downing, T. Hebron, B. Hanratty, E. Crowe, M. Doherty, J. Bray, and L. Lowry. The authors thank the families and staff of the psychiatric hospitals and units in Ireland and Northern Ireland who participated in the study and L. Eaves D.Sc., for comments on an earlier version of the manuscript.
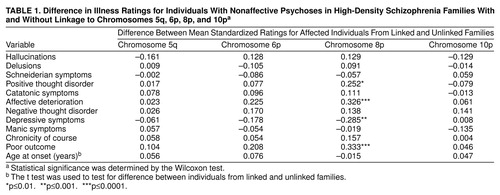 |

FIGURE 1. Two Hypothesized Models for the Relationship Between Four Different Disease Susceptibility Loci and Four Key Symptoms for a Diseasea
aModel A depicts a common pathway model in which all four genetic loci influence a common biological vulnerability, which in turn influences the occurrence of individual symptoms. Model B depicts an independent pathways model in which the four loci have direct and partially overlapping influences on specific disease symptoms. In this model, strong and weak relationships are indicated by, respectively, solid and dotted lines.

FIGURE 2. Mean Symptom and Outcome Scores of Individuals With Nonaffective Psychoses in Families With High-Density Schizophrenia Divided Into Fifths by Lod Score to Chromosome 8pa
aSymptom and outcome variables from the Major Symptoms of Schizophrenia Scale (31). The x axis displays the mean family lod scores (logarithm of the odds ratio for linkage) for families divided into fifths by lod scores; the y axis shows the mean symptom or course score for affected individuals in those families.
1. Defendorf AR: Clinical Psychiatry, a Text-Book for Students and Physicians Abstracted and Adapted from the Sixth German Edition of Kraepelin’s “Lehrbuch der Psychiatrie.” New York, Macmillan, 1902Google Scholar
2. Bleuler E: Dementia Praecox or the Group of Schizophrenias (1911). Translated by Zinkin J. New York, International Universities Press, 1950Google Scholar
3. Gottesman II: Schizophrenia Genesis: The Origins of Madness. New York, WH Freeman, 1990Google Scholar
4. Kendler KS: Schizophrenia: genetics, in Kaplan and Sadock’s Comprehensive Textbook of Psychiatry, 7th ed, vol 1. Edited by Sadock BJ, Sadock VA. Baltimore, Lippincott Williams & Wilkins, 1999, pp 1147–1159Google Scholar
5. Barr CL, Kennedy JL, Pakstis AJ, Wetterberg L, Sjogren B, Bierut L, Wadelius C, Wahlstrom J, Martinsson T, Giuffra L, Gelernter J, Hallmayer J, Moises HW, Kurth J, Cavalli-Sforza LL, Kidd KK: Progress in a genome scan for linkage in schizophrenia in a large Swedish kindred. Am J Med Genet 1994; 54:51–58Crossref, Medline, Google Scholar
6. Coon H, Jensen S, Holik J, Hoff M, Myles-Worsley M, Reimherr F, Wender P, Waldo M, Freedman R, Leppert M, Byerley W: Genomic scan for genes predisposing to schizophrenia. Am J Med Genet 1994; 54:59–71Crossref, Medline, Google Scholar
7. Levinson DF, Mahtani MM, Nancarrow DJ, Brown DM, Kruglyak L, Kirby A, Hayward NK, Crowe RR, Andreasen NC, Black DW, Silverman JM, Endicott J, Sharpe L, Mohs RC, Siever LJ, Walters MK, Lennon DP, Jones HL, Nertney DA, Daly MJ, Gladis M, Mowry BJ: Genome scan of schizophrenia. Am J Psychiatry 1998; 155:741–750Abstract, Google Scholar
8. Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR, Crow TJ, Sherrington R, DeLisi LE: A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet 1998; 81:364–376Crossref, Medline, Google Scholar
9. Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, Thornquist M, Ullrich G, McGrath J, Kasch L, Lamacz M, Thomas MG, Gehrig C, Radhakrishna U, Snyder SE, Balk KG, Neufeld K, Swartz KL, DeMarchi N, Papadimitriou GN, Dikeos DG, Stefanis CN, Chakravarti A, Childs B, Pulver AE: Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet 1998; 20:70–73Crossref, Medline, Google Scholar
10. Coon H, Myles-Worsley M, Tiobech J, Hoff M, Rosenthal J, Bennett P, Reimherr F, Wender P, Dale P, Polloi A, Byerley W: Evidence for a chromosome 2p13-14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol Psychiatry 1998; 3:521–527Crossref, Medline, Google Scholar
11. Straub RE, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D, Kendler KS: A potential vulnerability locus for schizophrenia on chromosome 6p24-22: evidence for genetic heterogeneity. Nat Genet 1995; 11:287–293Crossref, Medline, Google Scholar
12. Kendler KS, Tsuang MT: Nosology of paranoid schizophrenia and other paranoid psychoses. Schizophr Bull 1981; 7:594–610Crossref, Medline, Google Scholar
13. Kendler KS, Gruenberg AM, Tsuang MT: A family study of the subtypes of schizophrenia. Am J Psychiatry 1988; 145:57–62Link, Google Scholar
14. Kendler KS, McGuire M, Gruenberg AM, Walsh D: Outcome and family study of the subtypes of schizophrenia in the west of Ireland. Am J Psychiatry 1994; 151:849–856Link, Google Scholar
15. Leboyer M, Filteau MJ, Jay M, Campion D, d’Amato T, Guilloud-Bataille M, Hillaire D, Feingold J, des Lauriers A, Widlocher D: Clinical subtypes and age at onset in schizophrenic siblings. Psychiatry Res 1992; 41:107–114Crossref, Medline, Google Scholar
16. Slater E: Genetical causes of schizophrenic symptoms. Monatsschrift fur Psychiatrie und Neurologie 1947; 113:50–58Crossref, Medline, Google Scholar
17. Slater E: Psychotic and Neurotic Illnesses in Twins. London, Her Majesty’s Stationery Office, 1953Google Scholar
18. Bleuler M: The Schizophrenic Disorders: Long-Term Patient and Family Studies. New Haven, Conn, Yale University Press, 1978Google Scholar
19. DeLisi LE, Goldin LR, Maxwell ME, Kazuba DM, Gershon ES: Clinical features of illness in siblings with schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1987; 44:891–896Crossref, Medline, Google Scholar
20. Kendler KS, Karkowski-Shuman L, O’Neill FA, Straub RE, MacLean CJ, Walsh D: Resemblance of psychotic symptoms and syndromes in affected sibling pairs from the Irish Study of High-Density Schizophrenia Families: evidence for possible etiologic heterogeneity. Am J Psychiatry 1997; 154:191–198Link, Google Scholar
21. Brzustowicz LM, Honer WG, Chow EW, Hogan J, Hodgkinson K, Bassett AS: Use of a quantitative trait to map a locus associated with severity of positive symptoms in familial schizophrenia to chromosome 6p. Am J Hum Genet 1997; 61:1388–1396Google Scholar
22. Malhotra AK, Goldman D, Mazzanti C, Clifton A, Breier A, Pickar D: A functional serotonin transporter (5-HTT) polymorphism is associated with psychosis in neuroleptic-free schizophrenics. Mol Psychiatry 1998; 3:328–332Crossref, Medline, Google Scholar
23. Straub RE, MacLean CJ, O’Neill FA, Walsh D, Kendler KS: Support for a possible schizophrenia vulnerability locus in region 5q21-q31 in Irish families. Mol Psychiatry 1997; 2:148–155Crossref, Medline, Google Scholar
24. Kendler KS, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Easter SM, Webb BT, Zhang J, Walsh D, Straub RE: Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry 1996; 153:1534–1540Google Scholar
25. Straub RE, MacLean CJ, Martin RB, Ma Y, Myakishev MV, Harris-Kerr C, Webb BT, O’Neill FA, Walsh D, Kendler KS: A schizophrenia locus may be located in region 10p15-p11. Am J Med Genet 1998; 81:296–301Crossref, Medline, Google Scholar
26. Kendler KS, O’Neill FA, Burke J, Murphy B, Duke F, Straub RE, Shinkwin R, Ni Nuallain M, MacLean CJ, Walsh D: Irish study on high-density schizophrenia families: field methods and power to detect linkage. Am J Med Genet 1996; 67:179–190Crossref, Medline, Google Scholar
27. Kendler KS, McGuire M, Gruenberg AM, O’Hare A, Spellman M, Walsh D: The Roscommon Family Study, I: methods, diagnosis of probands, and risk of schizophrenia in relatives. Arch Gen Psychiatry 1993; 50:527–540Crossref, Medline, Google Scholar
28. Spitzer RL, Williams JBW, Gibbon M: Structured Clinical Interview for DSM-III-R (SCID). New York, New York State Psychiatric Institute, Biometrics Research, 1987Google Scholar
29. Kendler KS, Lieberman JA, Walsh D: The Structured Interview for Schizotypy (SIS): a preliminary report. Schizophr Bull 1989; 15:559–571Crossref, Medline, Google Scholar
30. Fleiss J: Statistical Methods for Rates and Proportions, 2nd ed. New York, John Wiley & Sons, 1981Google Scholar
31. Kendler KS, McGuire M, Gruenberg AM, Walsh D: An epidemiologic, clinical, and family study of simple schizophrenia in County Roscommon, Ireland. Am J Psychiatry 1994; 151:27–34Link, Google Scholar
32. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES: Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58:1347–1363Google Scholar
33. Conover WJ: Practical Nonparametric Statistics, 2nd ed. New York, John Wiley & Sons, 1980Google Scholar
34. Schwab SG, Eckstein GN, Hallmayer J, Lerer B, Albus M, Borrmann M, Lichtermann D, Ertl MA, Maier W, Wildenauer DB: Evidence suggestive of a locus on chromosome 5q31 contributing to susceptibility for schizophrenia in German and Israeli families by multipoint affected sib-pair linkage analysis. Mol Psychiatry 1997; 2:156–160Crossref, Medline, Google Scholar
35. Schwab SG, Albus M, Hallmayer J, Honig S, Borrmann M, Lichtermann D, Ebstein RP, Ackenheil M, Lerer B, Risch N, Maier W, Wildenauer DB: Evaluation of a susceptibility gene for schizophrenia on chromosome 6p by multipoint affected sib-pair linkage analysis. Nat Genet 1995; 11:325–327Crossref, Medline, Google Scholar
36. Maziade M, Bissonnette L, Rouillard E, Martinez M, Turgeon M, Charron L, Pouliot V, Boutin P, Cliche D, Dion C, Fournier JP, Garneau Y, Lavallee JC, Montgrain N, Nicole L, Pirè± A, Ponton AM, Potvin A, Wallot H, Roy MA, Mè±¥tte C (Le Groupe IREP):6p24-22 region and major psychoses in the Eastern Quebec population. Am J Med Genet 1997; 74:311–318Google Scholar
37. Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8: Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Am J Med Genet 1996; 67:580–594Crossref, Medline, Google Scholar
38. Faraone SV, Matise T, Svrakic D, Pepple J, Malaspina D, Suarez B, Hampe C, Zambuto CT, Schmitt K, Meyer J, Markel P, Lee H, Harkavy Friedman J, Kaufmann C, Cloninger CR, Tsuang MT: Genome scan of European-American schizophrenia pedigrees: results of the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet 1998; 81:290–295Crossref, Medline, Google Scholar
39. Schwab SG, Hallmayer J, Albus M, Lerer B, Hanses C, Kanyas K, Segman R, Borrman M, Dreikorn B, Lichtermann D, Rietschel M, Trixler M, Maier W, Wildenauer DB: Further evidence for a susceptibility locus on chromosome 10p14-p11 in 72 families with schizophrenia by nonparametric linkage analysis. Am J Med Genet 1998; 81:302–307Crossref, Medline, Google Scholar
40. Langfeldt G: The Prognosis in Schizophrenia and the Factors Influencing the Course of the Disease. Acta Psychiatr Neurol Scand Suppl 1937; 13Google Scholar
41. Zubin J, Sutton S, Salzinger K, Salzinger S, Burdock EI, Peretz D: A biometric approach to prognosis in schizophrenia, in Comparative Epidemiology of the Mental Disorders. Edited by Hoch PH, Zubin J. New York, Grune & Stratton, 1961, pp 143–203Google Scholar
42. Kendler KS, Karkowski LM, Walsh D: The structure of psychosis: latent class analysis of probands from the Roscommon Family Study. Arch Gen Psychiatry 1998; 55:492–499Crossref, Medline, Google Scholar
43. Cohen J: Statistical Power Analysis for the Behavioral Sciences. Orlando, Fla, Academic Press, 1977Google Scholar

