
Bipolar Affective Puerperal Psychosis: Genome-Wide Significant Evidence for Linkage to Chromosome 16
Abstract
Objective: Vulnerability to the triggering of bipolar episodes by childbirth aggregates in families and may define a genetically relevant subtype of bipolar disorder. The authors conducted a search by systematic whole genome linkage scan for loci influencing vulnerability to bipolar affective puerperal psychosis. Method: The authors selected families with bipolar disorder from their previous bipolar disorder genome scan, in which there was at least one family member with a manic or psychotic episode with an onset within 6 weeks of delivery. Individuals were coded as affected if they had been diagnosed with bipolar I disorder; bipolar II disorder; or schizoaffective disorder, bipolar type, according to DSM-IV. A total of 36 pedigrees contributed 54 affected sibling pairs to the cohort. A genome scan with 494 microsatellite markers was analyzed using GENEHUNTER and MAPMAKER/SIBS. Results: A genome-wide significant linkage signal was observed on chromosome 16p13, and a genome-wide suggestive linkage was observed on chromosome 8q24. No significant or suggestive linkage was observed in these regions in our original bipolar scan. Conclusions: This study identifies chromosomal regions that are likely to harbor genes that predispose individuals to bipolar affective puerperal psychosis. The identification of susceptibility genes would enhance understanding of pathogenesis and offer the possibility of improvements in treatment and risk prediction.
There is substantial evidence from family, twin, and adoption studies concerning the importance of genes in influencing susceptibility to bipolar disorder (1 , 2) . Progress in identifying the genetic variants that confer risk has improved in recent years, with replicated findings emerging from molecular genetic studies (3 – 6) . Despite this encouraging progress, however, findings have varied between studies, perhaps reflecting variation in the clinical and genetic characteristics of cohorts.
For complex genetic conditions such as bipolar disorder, there are marked benefits in focusing on a homogeneous clinical subtype, both to increase phenotypic and genetic homogeneity and to allow a specific subset of hypotheses to be tested (7) . In the case of bipolar disorder, a number of clinical subtypes have been investigated in molecular genetic studies, including rapid cycling illness, early age at onset, and lithium responsiveness. The present study focused on a clinical subtype of bipolar disorder that is defined by a vulnerability to the triggering of severe bipolar episodes by childbirth: bipolar affective puerperal (postpartum) psychosis. We have previously reported evidence that a vulnerability to postpartum episodes clusters in families (8 , 9) and episodes occurring in relationship to childbirth may be a marker for a more familial form of bipolar disorder (10) . A focus on bipolar disorder with a vulnerability to postpartum triggering is therefore a promising approach in the search for bipolar disorder susceptibility genes, and we have undertaken a systematic genome scan for loci that influence vulnerability to this subtype.
Method
Subject Recruitment
All subjects were Caucasian and of British or Irish origin and recruited in the United Kingdom and the Republic of Ireland through mental health services, patient support groups, and articles in the national media. Ethical approval was obtained prior to data collection, and written informed consent was obtained after a complete description of the study was given to each subject.
Assessment
All subjects were interviewed by a trained psychologist or psychiatrist using a semistructured research interview (Schedule for Clinical Assessment in Neuropsychiatry) (11) , and case note data were obtained. Consensus best estimate ratings of episode and lifetime diagnoses, according to DSM-IV criteria, were made by two independent investigators on the basis of all available clinical data. Any disagreements were rated by a third investigator and discussed in order to reach a consensus. Regular meetings were held between all interviewers and raters in order to maximize clinical consistency and reliability. Interrater reliability was assessed using clinical data from 20 cases (chosen to represent a typical cross section of subjects recruited within the study), which were rated by each investigator and compared against consensus to obtain individual kappa coefficients of reliability. Reliability was measured during the studies and shown to be excellent, with a mean kappa of 0.85 for DSM-IV diagnosis.
For female participants, information was obtained during interview and via case notes on the relationship of episodes of illness to childbirth. Consensus best estimate ratings were made for both the perinatal episode diagnosis and the timing of onset in relation to delivery by the methodology described previously with excellent reliability (mean kappa=0.90 and 0.93, respectively). This allowed us to identify the subset of female participants with a history of puerperal psychosis, defined in the present study as an episode of mania or psychosis with onset within 6 weeks of delivery. The 6-week onset was chosen as a compromise between very narrow (onset within 1 or 2 weeks) and very wide (onset within 6 months) definitions that have been used in previous studies on puerperal psychosis. The vast majority (95%) of women with an episode of puerperal psychosis in our cohort had an onset within 2 weeks of delivery. Additional details of the phenotypic assessment and diagnostic methods are provided elsewhere (12 – 14) .
Families were selected for inclusion in the current analysis according to the following criteria: 1) At least one female relative had a lifetime diagnosis that met DSM-IV criteria for bipolar I; bipolar II; or schizoaffective disorder, bipolar type, and had suffered an episode of mania or psychosis within 6 weeks of delivery; 2) at least one additional family member had an illness that met DSM-IV criteria for bipolar I; bipolar II; or schizoaffective disorder, bipolar type; and 3) the family was informative for linkage analysis.
Genotyping
The genome scan involved a two-stage design based on simulation work that showed that it was possible to achieve a similar power and type I error rate while considerably reducing the amount of genotyping. The strategy involved screening the stage 1 cohort with a 10 cM grid and then following up areas of interest with a tighter grid of markers every 5 cM. Seventeen regions that achieved nominally significant linkage in the full bipolar I disorder stage screen were taken forward into the second stage. In this study, both the first- and second-stage data were utilized in the analysis of the subset of families.
Laboratory work was undertaken using consistent methodologies in the molecular psychiatry laboratories at the University of Birmingham (Dr. Craddock), Cardiff University (Dr. Craddock), and the Psychiatric Genetics laboratory at Trinity College Dublin, Ireland, (Dr. Gill). The consistency and reliability of cross-center genotyping strategies were validated via a joint pilot study using markers on chromosome 21 (15) . All deoxyribonucleic acid (DNA) samples were either extracted from whole blood or from saline mouthwash samples using standard procedures. All polymerase chain reactions were performed on MJ Research Thermal Cyclers. Postpolymerase chain reaction—products from individual and multiplex reaction—were pooled in empirically determined ratios into size-specific marker sets prior to gel electrophoresis. This permitted up to 20 discrete marker loci to be analyzed in a single gel lane, with allele peak fluorescence intensities remaining within optimal limits (typically ∼200 to 4000 units). All markers were genotyped on either ABI377 XL DNA sequencers or ABI3100 sequencers using the software Genescan and Genotyper (Applied Biosystems, Foster City, Calif.). Further details of genotyping methodology are given in articles by Bennett et al. (13) and Lambert et al. (14) .
Statistical Analysis
Genetic relationships between family members were confirmed using marker data from across the genome and a suite of software packages: RELATIVE (16) , RELCHECK (17 , 18) , and PREST (19) . In-house software and the program GRR (20) were employed to detect monozygotic twins and cohort duplications to ensure that no one individual was typed in two different families. The presence of non-Mendelian errors was detected using the software PedCheck (21) .
Autosomal multipoint analyses were performed using the GENEHUNTER software package (22 , 23) , which calculates the maximum likelihood logarithm of the odds ratio (LOD) at each point in the genome using the maximum likelihood sharing probabilities (identical by descent) for each sibling pair. Maximum likelihood LOD scores for the X chromosome were estimated using the MAPMAKER/SIBS software package (24) . Marker allele frequencies were estimated using the program SPLINK (25) from our data set with maximum likelihood methods. Our method of analysis made no assumptions about the genetic model. The phenotypic model used was to define all individuals with a diagnosis of DSM-IV bipolar I; bipolar II; or schizoaffective disorder, bipolar type, as “affected.” All other individuals were considered as “phenotype unknown.”
We obtained empirical significance levels and the expected number of given LOD scores per genome screen by simulating 1,000 replicates of the entire data set under the null hypothesis of no linkage and then analyzing the data set with GENEHUNTER. These simulations maintained the same marker allele frequencies, marker locations, family structures, and individuals genotyped at each locus as in the observed data set. This accounted for the specific properties of the cohort studied and allowed for multiple testing.
Results
In the complete two-stage genome scan, 494 markers were genotyped in 232 bipolar families, with an average intermarker distance of 8.7 cM in stage 1 and 4.8 cM in the stage 2 regions (14) .
In the current analysis, which was limited to families containing a woman who had suffered an episode of postpartum psychosis, a total of 54 affected sibling pairs were included from 36 families (of which, 44 pairs from 26 families were included in stage 1). Table 1 shows the location and magnitude of the maximum linkage signal for each chromosome.
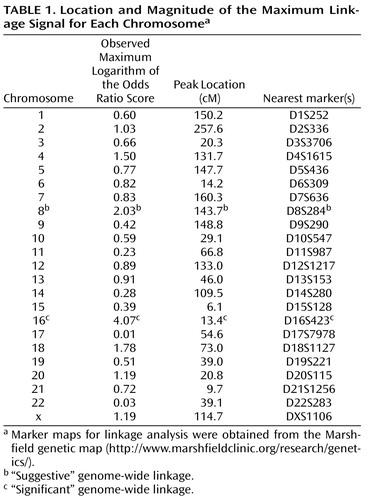
Simulation studies demonstrated that a maximum LOD of 3.62 was required for genome-wide significance according to the criteria by Lander and Kruglyak (26) . This equates to an LOD score of 3.62 or greater being obtained by chance (i.e., under the null hypothesis of no true linkage) only once in 20 genome scans. The maximum LOD of 4.07 observed on chromosome 16 was therefore genome-wide significant, with an empirical genome-wide significance p value of 0.02 ( Figure 1 ). The closest genotyped marker to our peak was DS16S423, and the LOD-1 interval (roughly corresponding to a 95% confidence interval) spanned approximately 9 cM (from genetic distance 10.4 cM [i.e., D16S423 the most telomeric marker typed] to 19.4 cM). The estimated probability of allele sharing between affected siblings at the peak was 0.75, a substantial elevation above the null expectation of 0.50.
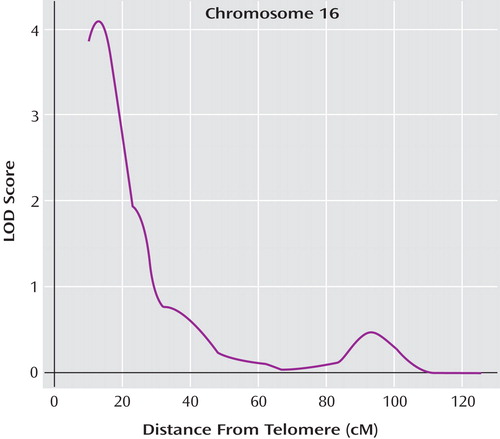
a The map distances were obtained from the Marshfield genetic map (http://www.marshfieldclinic.org/research/genetics/).
An LOD of 1.95 or higher was expected to occur by chance once per genome scan (i.e., genome-wide “suggestive” linkage according to the criteria by Lander and Kruglyak [26] ). In addition to the genome-wide significant region on chromosome 16, we observed a genome-wide suggestive region on chromosome 8q24, with an LOD score of 2.03 close to marker D8S284 and an LOD-1 interval of 14 cM (from 135.7 cM to 149.7 cM) ( Figure 2 ). The estimated probability of allele sharing between affected siblings at this peak was 0.69, which was again a substantial elevation above the null expectation of 0.50.
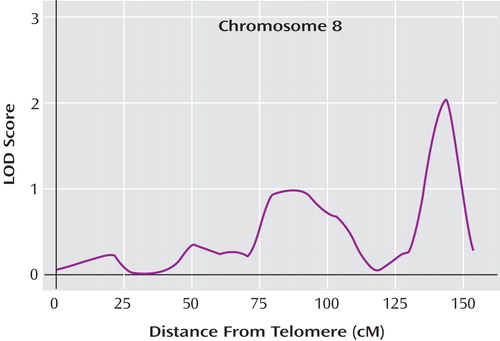
a The map distances were obtained from the Marshfield genetic map (http://www.marshfieldclinic.org/research/genetics/).
Discussion
Linkage studies in bipolar disorder have tended to focus on bipolar I disorder alone or have extended the phenotype to include related diagnoses such as bipolar II disorder, recurrent major depression, and schizoaffective disorder. It is highly likely, however, that the bipolar diagnostic label covers a heterogeneous collection of conditions with differing etiological factors. Increasing attention is now being given to clinical subtypes of the disorder that may represent more homogeneous forms of the illness.
In this study, we have focused on a clinical subtype defined by a vulnerability to severe episodes of illness being triggered by childbirth. We have previously reported 1) compelling evidence that a vulnerability to postpartum bipolar episodes clusters in families (8) and 2) data suggesting that a vulnerability to puerperal episodes is a marker for a more familial form of bipolar disorder (10) . Additionally, we have reported a number of candidate gene association studies in women with puerperal psychosis (27) and have shown suggestive, although preliminary, evidence for an association between puerperal psychosis and a variant at the serotonin transporter gene on chromosome 17 (28) . In the present study, we employed the complementary, positional approach of a genome-wide linkage study in sibling pairs, focusing on those families in which a female proband has suffered an episode of mania or psychosis within 6 weeks of delivery.
We found one chromosome region (16p13) with an LOD score (4.07) meeting the Lander and Kruglyak criteria (26) for genome-wide significance (p=0.02). A finding of this magnitude would therefore be expected by chance only once in every 43 genome scans conducted with similar family pedigrees and markers. We also report an additional chromosomal region (8q24) that meets the Lander and Kruglyak (26) criteria for genome-wide suggestive linkage.
Previous Findings at Loci 16p13 and 8q24
Although, to our knowledge, this is the first linkage study to focus on families containing cases of bipolar affective puerperal psychosis, it is of interest to compare the location of these chromosomal regions with those previously reported in linkage studies of bipolar disorder families unselected for onset in the puerperium. Although the two loci did not emerge from our own genome scan of bipolar disorder (13 , 14) , there have been previous reports of linkage in these regions.
16p13
The 16p13 region has not been implicated in meta-analyses of bipolar linkage scans (3 , 4 , 29) , but a number of individual studies have reported evidence of linkage in this chromosomal location. Ewald et al. (30) reported an LOD score of 2.5 in two Danish pedigrees under a recessive model; their peak was less than 3 cM from our own and lay within our LOD-1 region. Other studies have also reported linkage in this general region. McInnes et al. (31) reported modest evidence for linkage (LOD=1.46) in two Costa Rican pedigrees, with a peak 12 cM telomeric from ours. Finally, multiple waves of the National Institute of Mental Health (NIMH) Genetics Initiative on Bipolar Disorder reported evidence for linkage in this region, with LOD scores >2 across a 17 cM region beginning 10 cM centromeric from our peak (32 – 34) . Most recently, the consortium has reported that the evidence for linkage in this region is substantially increased (LOD score=4.5) when employing mania at onset as a covariate in the analysis (35) .
The location of the peak of our genome-wide significant linkage signal on chromosome 16p13, therefore, coincides with a region that has been associated with bipolar disorder in previous linkage studies. It is possible that the families in the previous studies were particularly susceptible to postpartum triggering of illness. In the NIMH study, for example, it has been reported that almost 50% of the female bipolar probands reported episodes of severe illness in relation to childbirth (36) . However, it is also possible that the short arm of chromosome 16 contains two or more loci harboring vulnerability genes for various forms of bipolar disorder.
8q24
The 8q24 region has also been implicated in previous linkage studies. Our peak at 143 cM was flanked by two previously reported linkage signals. Cichon et al. (37) reported an LOD score of 3.62 in 75 families that peaked at 130 cM, and 65 pedigrees were reported by the Johns Hopkins group (38 , 39) , with a maximum LOD score of 3.32 that peaked at 148 cM, the latter falling within our LOD-1 region. Although two of the published meta-analyses of bipolar linkage scans (3 , 4) did not implicate this region of chromosome 8, a recent pooled-data analysis proved to be more germane at this locus (29) . This analysis included 11 genome scans covering 5,179 individuals from 1,067 families and benefited from combining the original genotype data rather than the linkage statistics or p values. Employing a “broad” affected model that included both bipolar I and II probands, the study found evidence for genome-wide significant linkage (LOD=3.40) in this region with a peak at 151 cM.
Limitations
Despite the encouraging findings reported in this study, they must be interpreted in the light of a number of limitations.
Cohort size
In the context of studies on complex disorders, our cohort size was relatively modest (54 sibling pairs). The fact that such a cohort could yield one signal that is genome-wide significant and another suggestive signal suggests that our study has indeed benefited from increasing genetic homogeneity and illustrates the point that carefully selected cohorts of modest size may assist in finding genes for bipolar disorder. With this cohort size, however, genes conferring a small or modest increase in vulnerability to illness are unlikely to be identified. Further, it is desirable that our findings are replicated in independent cohorts, although this may require a number of further scans before it will become clear whether the loci reported in the present study actually contain vulnerability genes for bipolar affective puerperal psychosis.
Biological model
It is worth considering the model of puerperal triggering of bipolar episodes that underlies the current analysis. We consider bipolar disorder to represent an etiologically heterogeneous collection of disorders, including a form in which women are vulnerable to the triggering of episodes by childbirth. In this model, the genetic variants influencing vulnerability to puerperal triggering are the same variants that influence vulnerability to bipolar disorder. By selecting families through an index case of bipolar affective puerperal psychosis, we have sought to identify a more homogeneous cohort, which despite being smaller in number will facilitate the search for genes influencing disease susceptibility. An alternative model is that the trigger genes are independent of the bipolar genes and may be considered additional course modifying factors. In order to identify such course modifying genes we would need to study families multiply affected with puerperal psychosis. In the present study, we have identified families with an index proband that has suffered from puerperal psychosis; however, the proband may be the only individual in the family to suffer with a postpartum episode. The number of families in which there were two or more women with puerperal psychosis was prohibitively small for meaningful analysis of puerperal psychosis sibling pairs alone. Further recruitment of families multiply affected with perinatal mood disorder would be beneficial and allow an exploration of the alternative, course modifying model of postpartum triggering.
Specificity
We selected families for inclusion in this study based upon the occurrence of an episode of postpartum psychosis. There was no evidence of systematic differences between illness in the nonpuerperal relatives in these families and other families in our sib pair cohort. However, it remains possible that there is one or more correlated characteristics that accounts for the linkage obtained. Future linkage studies examining a range of clinical variables as covariates may prove beneficial in refining more heterogeneous bipolar disorder phenotypes.
Gene identification
The regions of interest we have identified contain many hundreds of genes. More extensive investigation is needed to narrow the regions of interest and to examine the potential candidate genes they contain. There are, however, a number of potentially relevant genes in these regions, including ABAT and GRIN2A (chromosome 16) and ADCY8 (chromosome 8) that will benefit from further study.
In summary, we report the first systematic genome scan aimed at localizing genes that influence susceptibility to bipolar affective puerperal psychosis and provide further support for the hypothesis that this is a genetically meaningful subtype of bipolar disorder. We have identified regions of interest on chromosomes 16p13 (LOD=4.07, genome-wide significant) and 8q24 (LOD=2.03, genome-wide suggestive). Our linkage findings point to the benefits of focusing on the subset of bipolar disorder in which women have a vulnerability to the triggering of episodes by childbirth and provide strong evidence for a susceptibility locus on chromosome 16. Considerable investigation is required to identify the genetic variant or variants involved, but finding genes that influence susceptibility to the postpartum triggering of bipolar episodes will, it is hoped, lead to a better understanding of the etiology of these conditions and to the development of better treatment and prevention for women who suffer such devastating episodes during this time.
1. Craddock N, Jones I: Genetics of bipolar disorder. J Med Gen 1999; 36:585–594Google Scholar
2. Jones I, Craddock N: Do puerperal psychotic episodes identify a more familial subtype of bipolar disorder? results of a family history study. Psychiatr Genet 2002; 12:177–180Google Scholar
3. Badner JA, Gershon ES: Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatr 2002; 7:405–411Google Scholar
4. Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI Jr, Craddock N, DePaulo JR, Baron M, Gershon ES, Ekholm J, Cichon S, Turecki G, Claes S, Kelsoe JR, Schofield PR, Badenhop RF, Morissette J, Coon H, Blackwood D, McInnes LA, Foroud T, Edenberg HJ, Reich T, Rice JP, Goate A, McInnis MG, McMahon FJ, Badner JA, Goldin LR, Bennett P, Willour VL, Zandi PP, Liu J, Gilliam C, Juo SH, Berrettini WH, Yoshikawa T, Peltonen L, Lonnqvist J, Nothen MM, Schumacher J, Windemuth C, Rietschel M, Propping P, Maier W, Alda M, Grof P, Rouleau GA, Del-Favero J, Van Broeckhoven C, Mendlewicz J, Adolfsson R, Spence MA, Luebbert H, Adams LJ, Donald JA, Mitchell PB, Barden N, Shink E, Byerley W, Muir W, Visscher PM, Macgregor S, Gurling H, Kalsi G, McQuillin A, Escamilla MA, Reus VI, Leon P, Freimer NB, Ewald H, Kruse TA, Mors O, Radhakrishna U, Blouin JL, Antonarakis SE, Akarsu N: Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: bipolar disorder. Am J Hum Genet 2003; 73:49–62Google Scholar
5. DePaulo JR Jr: Genetics of bipolar disorder: where do we stand? Am J Psychiatry 2004; 161:595–597Google Scholar
6. Craddock N, O’Donovan MC, Owen MJ: The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet 2005; 42:193–204Google Scholar
7. Lander ES, Schork NJ: Genetic dissection of complex traits. Science 1994; 265:2037–2048Google Scholar
8. Jones I, Craddock N: Familiality of the puerperal trigger in bipolar disorder: results of a family study. Am J Psychiatry 2001; 158:913–917Google Scholar
9. Forty L, Jones L, Macgregor S, Caesar S, Cooper C, Hough A, Dean L, Dave S, Farmer A, McGuffin P, Brewster S, Craddock N, Jones I: Familiality of postpartum depression in unipolar disorder: results of a family study. Am J Psychiatry 2006; 163:1549–1553Google Scholar
10. Jones I, Craddock N: Do puerperal psychotic episodes identify a more familial subtype of bipolar disorder? results of a family history study. Psychiatr Genet 2002; 12:177–180Google Scholar
11. Wing JK, Babor T, Brugha T, Burke J, Cooper JE, Giel R: Scan: Schedules for Clinical Assessment in Neuropsychiatry. Arch Gen Psychiatry 1990; 47:589–593Google Scholar
12. Robertson E, Jones I, Benjamin J, Murdoch C, Pelios G, Brockington I, Craddock N: Approaches to the ascertainment, recruitment and clinical assessment of women with puerperal psychosis. Arch Womens Ment Health 2000; 3:59–64Google Scholar
13. Bennett P, Segurado R, Jones I, Bort S, McCandless F, Lambert D, Heron J, Comerford C, Middle F, Corvin A, Pelios G, Kirov G, Larsen B, Mulcahy T, Williams N, O’Connell R, O’Mahony E, Payne A, Owen M, Holmans P, Craddock N, Gill M: The Wellcome Trust UK-Irish Bipolar Affective Disorder Sibling-Pair Genome Screen: first stage report. Mol Psychiatry 2001; 7:189–200Google Scholar
14. Lambert D, Middle F, Hamshere ML, Segurado R, Raybould R, Corvin A, Green E, O’Mahony E, Nikolov I, Mulcahy T, Haque S, Bort S, Bennett P, Norton N, Owen MJ, Kirov G, Lendon C, Jones L, Jones I, Holmans P, Gill M, Craddock N: Stage 2 of the Wellcome Trust UK-Irish Bipolar Affective Disorder Sibling-Pair Genome Screen: evidence for linkage on chromosomes 6q16-q21, 4q12-q21, 9p21, 10p14-p12 and 18q22. Mol Psychiatry 2005; 10:831–841Google Scholar
15. Bennett P, Mulcahy T, Owen MJ, Craddock N, Gill M: The Wellcome Trust UK-Irish Bipolar Sib-Pair Study: chromosome 21. Am J Med Genet 1998; 81:541Google Scholar
16. Göring HH, Ott J: Relationship estimation in affected sib-pair analysis of late-onset diseases. Eur J Hum Genet 1997; 5:69–77Google Scholar
17. Broman KW, Weber JL: Estimation of pairwise relationships in the presence of genotyping errors. Am J Hum Genet 1998; 63:1563–1564Google Scholar
18. Boehnke M, Cox NJ: Accurate inference of relationships in sib-pair linkage studies. Am J Hum Genet 1997; 61:423–429Google Scholar
19. McPeek MS, Sun L: Statistical tests for detection of misspecified relationships by use of genome-screen data. Am J Hum Genet 2000; 66:1076–1094Google Scholar
20. Abecasis GR, Cherny SS, Cookson WOC, Cardon LR: GRR: graphical representation of relationship errors. Bioinformatics 2001; 17:742–743Google Scholar
21. O’Connell JR, Weeks DE: PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63:259–266Google Scholar
22. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES: Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58:1347–1363Google Scholar
23. Kruglyak L, Lander ES: Faster multipoint linkage analysis using Fourier transforms. J Comput Biol 1998; 5:1–7Google Scholar
24. Kruglyak L, Lander ES: Complete multipoint sib pair analysis of qualitative and quantitative traits. Am J Hum Genet 1995; 57:439–454Google Scholar
25. Holmans P, Clayton D: Efficiency of typing unaffected relatives in an affected-sib-pair linkage study with single-locus and multiple tightly linked markers. Am J Hum Genet 1995; 57:1221–1232Google Scholar
26. Lander E, Kruglyak L: Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247, 1995Google Scholar
27. Robertson E, Jones I, Middle F, Moray J, Craddock N: No association between two polymorphisms at the 5HT2A gene and bipolar affective puerperal psychosis. Acta Psychiatr Scand 2003; 108:387–391Google Scholar
28. Coyle N, Jones I, Roberston E, Lendon C, Craddock N: Variation at the serotonin transporter gene influences susceptibility to bipolar affective puerperal psychosis. Lancet 2000; 356:1490–1491Google Scholar
29. McQueen MB, Devlin B, Faraone SV, Nimgaonkar VL, Sklar P, Smoller JW, Abou Jamra R, Albus M, Bacanu SA, Baron M, Barrett TB, Berrettini W, Blacker D, Byerley W, Cichon S, Coryell W, Craddock N, Daly MJ, Depaulo JR, Edenberg HJ, Foroud T, Gill M, Gilliam TC, Hamshere M, Jones I, Jones L, Juo SH, Kelsoe JR, Lambert D, Lange C, Lerer B, Liu J, Maier W, Mackinnon JD, McInnis MG, McMahon FJ, Murphy DL, Nothen MM, Nurnberger JI, Pato CN, Pato MT, Potash JB, Propping P, Pulver AE, Rice JP, Rietschel M, Scheftner W, Schumacher J, Segurado R, Van Steen K, Xie W, Zandi PP, Laird NM: Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet 2005; 77:582–595Google Scholar
30. Ewald H, Flint T, Kruse TA, Mors O: A genome-wide scan shows significant linkage between bipolar disorder and chromosome 12q24.3 and suggestive linkage to chromosomes 1p22–21, 4p16, 6q14–22, 10q26 and 16p13.3. Mol Psychiatry 2002; 7:734–744Google Scholar
31. McInnes LA, Escamilla MA, Service SK, Reus VI, Leon P, Silva S, Rojas E, Spesny M, Baharloo S, Blankenship K, Peterson A, Tyler D, Shimayoshi N, Tobey C, Batki S, Vinogradov S, Meza L, Gallegos A, Fournier E, Smith LB, Barondes SH, Sandkuijl LA, Freimer NB: A complete genome screen for genes predisposing to severe bipolar disorder in two Costa Rican pedigrees. Proc Natl Acad Sci U S A 1996; 93:13060–13065Google Scholar
32. Edenberg HJ, Foroud T, Conneally PM, Sorbel JJ, Carr K, Crose C, Willig C, Zhao J, Miller M, Bowman E, Mayeda A, Rau NL, Smiley C, Rice JP, Goate A, Reich T, Stine OC, McMahon F, DePaulo JR, Meyers D, Detera-Wadleigh SD, Goldin LR, Gershon ES, Blehar MC, Nurnberger JI Jr: Initial genomic scan of the NIMH genetics initiative bipolar pedigrees: chromosomes 3, 5, 15, 16, 17, and 22. Am J Med Genet 1997; 74:238–246Google Scholar
33. Dick DM, Foroud T, Edenberg HJ, Miller M, Bowman E, Rau NL, DePaulo JR, McInnis M, Gershon E, McMahon F, Rice JP, Bierut LJ, Reich T, Nurnberger J Jr. Apparent replication of suggestive linkage on chromosome 16 in the NIMH genetics initiative bipolar pedigrees. Am J Med Genet 2002; 114:407–412Google Scholar
34. McMahon FJ, Austin L, Steele CJM, Foroud T, Meyer ET, Chen YS, Cox NJ, Nurnberger JI, NIMH Genetics Initiative Bipolar Disorder Group. Genome-wide linkage analysis of the NIMH Genetics Initiative Wave 4 Bipolar Disorder Pedigrees. Am J Med Genet (Neuropsychiatric Genetics) 2004; 130B:1–180Google Scholar
35. Kassem L, Lopez V, Hedeker D, Steele J, Zandi P, Bipolar Disorder Consortium NIMH Genetics Initiative; McMahon FJ: Familiality of polarity at illness onset in bipolar affective disorder. Am J Psychiatry 2006; 163:1754–1759Google Scholar
36. Blehar MC, DePaulo JR, Gershon ES, Reich T, Simpson SG, Nurenberger JI: Women with bipolar disorder: findings from the NIMH Genetics Initiative Sample. Psychopharmacol Bull 1998; 34:239–243Google Scholar
37. Cichon S, Schumacher J, Muller DJ, Hurter M, Windemuth C, Strauch K, Hemmer S, Schulze TG, Schmidt-Wolf G, Albus M, Borrmann-Hassenbach M, Franzek E, Lanczik M, Fritze J, Kreiner R, Reuner U, Weigelt B, Minges J, Lichtermann D, Lerer B, Kanyas K, Baur MP, Wienker TF, Maier W, Rietschel M, Propping P, Nothen MM: A genome screen for genes predisposing to bipolar affective disorder detects a new susceptibility locus on 8q. Hum Mol Genet 2001; 10:2933–2944Google Scholar
38. McInnis MG, Dick DM, Willour VL, Avramopoulos D, MacKinnon DF, Simpson SG, Potash JB, Edenberg HJ, Bowman ES, McMahon FJ, Smiley C, Chellis JL, Huo Y, Diggs T, Meyer ET, Miller M, Matteini AT, Rau NL, DePaulo JR, Gershon ES, Badner JA, Rice JP, Goate AM, Detera-Wadleigh SD, Nurnberger JI, Reich T, Zandi PP, Foroud TM: Genome-wide scan and conditional analysis in bipolar disorder: evidence for genomic interaction in the National Institute of Mental Health Genetics Initiative Bipolar Pedigrees. Biol Psychiatry 2003; 54:1265–1273Google Scholar
39. Avramopoulos D, Willour VL, Zandi PP, Huo Y, MacKinnon DF, Potash JB, DePaulo JR Jr, McInnis MG: Linkage of bipolar affective disorder on chromosome 8q24: follow-up and parametric analysis. Mol Psychiatry 2004; 9:191–196Google Scholar

