
Association of a Functional Deficit of the BK Ca Channel, a Synaptic Regulator of Neuronal Excitability, With Autism and Mental Retardation
Abstract
Objective: Autism is a complex, largely genetic psychiatric disorder. In the majority of cases, the cause of autism is not known, but there is strong evidence for a genetic etiology. To identify candidate genes, the physical mapping of balanced chromosomal aberrations is a powerful strategy, since several genes have been characterized in numerous disorders. In this study, the authors analyzed a balanced reciprocal translocation arising de novo in a subject with autism and mental retardation. Method: The authors performed the physical mapping of the balanced 9q23/10q22 translocation by fluorescent in situ hybridization experiments using bacterial artificial chromosome clones covering the areas of interest. Results: Findings revealed that the KCNMA1 gene, which encodes the alpha-subunit of the large conductance Ca 2+ -activated K + (BK Ca ) channel, a synaptic regulator of neuronal excitability, is physically disrupted. Further molecular and functional analyses showed the haploinsufficiency of this gene as well as decreased activity of the coded BK Ca channel. This activity can be enhanced in vitro by addition of a BK Ca channel opener (BMS-204352). Further mutational analyses on 116 autistic subjects led to the identification of an amino acid substitution located in a highly conserved domain of the protein not found in comparison subjects. Conclusions: These results suggest a possible association between a functional defect of the BK Ca channel and autistic disorder and raise the hypothesis that deficits in synaptic transmission may contribute to the physiopathology of autism and mental deficiency.
Autism (MIM 209850) is a neurodevelopmental disorder beginning before the age of 3 that is characterized by impaired reciprocal social interaction and communication as well as restricted and stereotyped patterns of interests and activities. The prevalence of autism is estimated to be at least 10 per 10,000, with a male to female ratio of approximately 4:1 (1) . In the majority of cases, the cause of autism is unknown, but many studies, including correlation in twins and association of autism with Mendelian diseases, support a genetic etiology for this condition (2 , 3) . Autistic disorder appears to be clinically and genetically heterogeneous, suggesting that multiple loci are involved (4) . It is therefore suggested that numerous chromosomal loci contribute to the genetic susceptibility to the disorder, since the identification of chromosomal abnormalities in autistic individuals and data from large-scale linkage studies suggest a linkage to several different chromosomal regions (2 , 5) . Recently, mutations in the NLGN3 and NLGN4 genes have been associated with autism, Asperger syndrome, and X-linked mental retardation (6 , 7) , which suggests, in these cases, that autistic spectrum disorders follow a recessive Mendelian mode of transmission.
The incidence of chromosomal aberrations in the population of patients with autism is much higher than would be expected relative to low incidence in the healthy population, suggesting that there is a causal relationship between the occurrence of autistic disorder and the chromosomal anomaly.
In this study, we describe a complete molecular genetic and functional analysis of an autistic disorder patient with a de novo balanced 9q23/10q22 translocation. We show that the disruption of the KCNMA1 gene leads to a haploinsufficiency and a decreased activity of the KCNMA1 (or BK Ca ) protein. We then demonstrate that the action of a BK Ca channel opener is able to enhance a BK Ca current. These findings support the hypothesis that autism is associated with defects in neuronal circuitry and excitability.
Method
After a complete description of the study was given, written informed consent from the parents was obtained as well as assent from the subjects.
Clinical Data for Autistic Subjects
Patient carrying the de novo 9q23/10q22 translocation
The patient was a 6-year-old boy at the time of clinical evaluation. He had impairments of reciprocal social interactions and communication skills, lack of spoken language, and poor communicating gestures. He also displayed restricted stereotyped behaviors. The diagnosis of autistic disorder was confirmed by administration of the Autism Diagnostic Interview–Revised (8) and the Childhood Autism Rating Scale (9) . The patient satisfied the prespecified cutoff scores in all three of the following symptom areas of the Autism Diagnostic Interview–Revised: qualitative impairment, with a reciprocal social interaction score of 30 (cutoff for autism=10); qualitative impairment, with a nonverbal communication score of 13 (cutoff for autism=7); and repetitive behaviors and stereotyped patterns score of 7 (cutoff for autism=3). In addition, he showed evidence of first symptoms at <36 months old. His score of 40 for the Childhood Autism Rating Scale evaluation fell within the severe range of autism. He met all DSM–IV criteria for autistic disorder. Moreover, the results of the Psychoeducational Profile–Revised indicated a developmental score equivalent to an age of 23 months (10) and confirmed the associated diagnosis of severe mental retardation according to DSM–IV criteria. Physical examinations revealed normal growth parameters, and there were no discernible dysmorphic features. Electroencephalogram and magnetic resonance imaging findings were normal. The patient’s parents were nonconsanguineous and free of any medical and neuropsychiatric disorders. His 8-year-old brother was positive for phenylketonuria and had been treated with a phenylalanine-restricted diet from birth and later developed specific language impairment without mental deficiency. Analysis of the patient’s karyotype revealed no chromosomal disorders. In addition, no abnormalities of phenylalanine or tetrahydrobiopterin levels (phenylalanine hydroxylase cofactor) were detected.
The analysis of the karyotype of the patient and his parents revealed a de novo balanced translocation (46, XY, t [9; 10] [q23;q22]) associated with autistic disorder and severe mental retardation.
Patient carrying the ALA138VAL substitution
The patient was the fourth-born boy of a Tunisian family. He had impairments of reciprocal social interactions and communication skills, lack of spoken language, and poor communicating gestures. He also displayed restricted stereotyped behaviors. The diagnosis of autistic disorder was confirmed by the following: administration of the Autism Diagnostic Interview–Revised (with qualitative impairment), with a reciprocal social interaction score of 34 (cutoff for autism=10); qualitative impairment, with a nonverbal communication score of 15 (cutoff for autism=7); and repetitive behaviors and stereotyped patterns score of 17 (cutoff for autism=3). The patient showed evidence of first symptoms at <36 months old. He met all DSM–IV criteria for autistic disorder, and he had severe mental retardation and seizures. In addition, he had discrete facial dysmorphia with long face and retrognathism, but parental description was not available. Computed tomography (CT) scan results were normal. The child’s parents were nonconsanguineous and free of any medical and neuropsychiatric disorders.
Fluorescence in Situ Hybridization
Fluorescence in situ hybridization analyses were performed on metaphase spreads that were obtained from peripheral white blood cells from an Epstein-Barr virus immortalized cell line from the patient. Selected bacterial artificial chromosome/P1 artificial chromosome clones were obtained from the Children’s Hospital Oakland Research Institute and were biotynilated by nick-translation using the BioNick labeling system (Life Technologies, Gaithersburg, Md.). To identify chromosomes 9 and 10 on the metaphases spreads, we used alpha-satellite or telomeric probes (Qbiogen, Illkirch, France).
Semiquantitative Reverse Transcription-Polymerase Chain Reaction
We used total ribonucleic acid (RNA) extracted from Epstein-Barr virus-transformed lymphoblastoid cells for reverse transcription–polymerase chain reaction according to standard procedures. We amplified a fragment of the KCNMA1 C-deoxyribonucleic acid (cDNA) with a forward primer in exon 9 (5′-GGCTCCTATAGTGCGGTTAGT-3′) and a reverse primer in exon 14 (5′-TGTTTAGCAGATGGGCCTTGTT-3′). In addition, we coamplified a cDNA fragment of the β -actin gene as an internal standard with the following primers: forward primer, 5′-TCATGCCATCCTGCGTCTGGACCT-3′ and reverse primer, 5′-CCGGACTCATCGTACTCCTGCTTG-3′, with an annealing temperature of 60 °C.
Electrophysiology
Whole-cell recordings of K + currents were acquired in lymphoblastoid cell lines obtained from the autistic patients and comparison subjects. The cells were washed and spun three times and suspended in a physiological saline solution composed of the following (in mmol/liter): NaCl=140; KCl=4; CaCl 2 =2; MgCl 2 =1; NaH 2 PO 4 =0.33; glucose=11.1; Hepes buffer=10; pH balanced to 7.4 with NaOH. An aliquot of these cells was then placed in a Petri dish containing 2 ml physiological saline solution. Petri dishes with cells were placed on the stage of an Elipse TE-300 Nikon microscope. The cells were superfused with experimental solutions via a parallel pipe system lowered into the vicinity of the cells. Solution exchange around each patched cell was estimated to be less than 10s. Cells were intracellularly perfused with a 400 nM free Ca 2+ pipette solution (to activate the BK Ca ) containing (in mmol/liter) K-glutamate=125; KCl=20; CaCl 2 =0.7; Mg-ATP=1; Mg-GTP=1; EGTA=1; Hepes buffer=10; pH balanced to 7.2 with KOH. Pipette tip resistance ranged between 3 MΩ and 5MΩ.. Macroscopic K + currents were generated by progressive 8mV depolarizing steps (500 msec duration, 5 second intervals) from a constant holding potential of –70 mV. BK Ca currents were defined as the outward current inhibited by 100 nmol/liter iberiotoxin (IbTx), a blocker of BK Ca channels (11) . The capacitance of each cell was estimated by integrating the capacitive current that was generated by a 10 mV hyperpolarizing pulse after electronic cancellation of pipette-patch capacitance that was provided by the Axopatch 200B amplifier (Axon Instruments, Union City, Calif.). IbTx-sensitive currents were expressed as current density (pA/pF). Voltage clamp protocol and data acquisition were controlled with pClamp V 8.1 software (Axon Instruments). Data were digitized at 20 kHz and recorded with an 80486 computer after filtering with a five-pole Bessel filter at 2 kHz. All experiments were conducted at room temperature. All data are expressed as means and standard deviations. Statistical comparisons between groups were performed with two-way repeated measures analysis of variance with a subsequent Dunnett post hoc analysis test. Significance was set at p<0.05.
Mutation Analysis
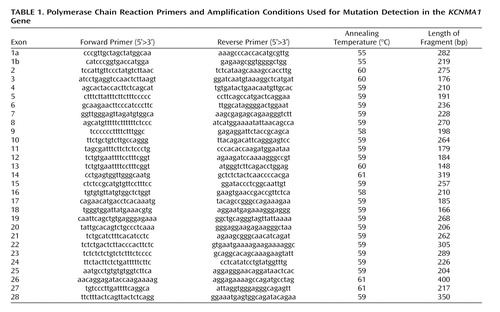
Polymerase chain reactions were performed in 50-μL reaction volumes containing 100 ng of the genomic DNA of the patients (10 pM of each primer, 1.25 mM of dNTPs, 0.5 U Taq DNA polymerase [Promega], and 1.5 mM MgCl 2 ). An initial denaturation of 5 minutes at 94°C was followed by 30 1-minute cycles at 94°C, 1 minute of annealing, a 1-minute extension at 72°C, and a final extension step of 7 minutes at 72°C (see Table 1 for polymerase chain reaction conditions and primers sequence). The products were then checked on a 1.5% agarose gel to verify amplification prior to analysis by denaturing high-performance liquid chromatography using the WAVE 3500 HT system (Transgenomic, Inc., Omaha, Neb.). For each amplified exon, melting profiles and temperatures were predicted by the Transgenomic Navigator software version 1.5.1. Pairs of amplified fragments from patient and comparison subject polymerase chain reaction products were pooled, denatured at 95°C for 3 minutes, and cooled to 40°C in decreasing increments of 0.05°C per second. The products were then injected and eluted with an acetonitrile gradient at a flow rate of 1.5 ml/min, with a mobile phase composed of two buffers (buffer A: 0.1 M triethylammonium acetate; buffer B: 0.1 M triethylammonium acetate with 25% acetonitrile). For each sample pair that exhibited an abnormal elution profile, the polymerase chain reaction products were purified and sequenced on an ABI377 DNA sequencer (Perkin Elmer).
Results
Molecular Analysis
Fluorescent in situ hybridization studies with bacterial artificial chromosome clones obtained from the Children’s Hospital Oakland Research Institute library enabled us to map the 9q23 breakpoint to RP11-85P23 (accession number AL161632) and the 10q22 breakpoint to RP11-619F23 (accession number AL627447) (part A of Figure 1 ). Sequence analysis of the two breakpoint areas revealed 1) the absence of a gene in the 9q23 region and 2) localization of the KCNMA1 gene (NCBI NM_002227, MIM number [*600150]), encoding for a large conductance calcium-activated K + (BK Ca ) channel in the 10q22 region (part B of Figure 1 ) (12) . Polymerase chain reaction analyses on the RP11-619F23 clone indicated that the 10q breakpoint was located in the first intron. Molecular analyses of the genomic sequence spanning the two breakpoints revealed a 14 bp deletion (part C of Figure 1 ) that is located in large noncoding regions.
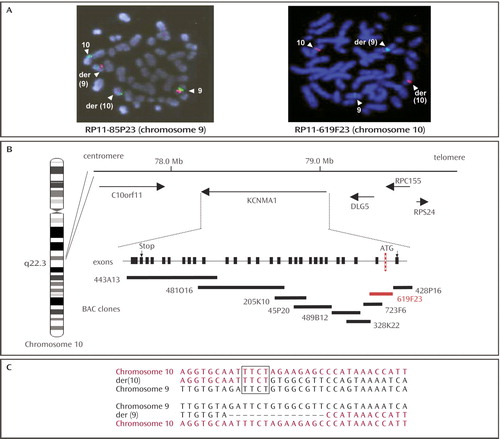
a In part A, metaphase spreads from the patient with the 9/10 translocation showing red fluorescence in situ hybridization signals obtained with RP11-619F23 (chromosome 10), with centromere-specific probes (green) (Qbiogen, Illkirch, France). The hybridization signals (white arrowheads) on both the der(9) and der(10) chromosomes indicate that the bacterial artificial chromosome clones spanned the breakpoints. Part B shows localization of the 10q breakpoint and schematic representation of the bacterial artificial chromosome contig along the KCNMA1 gene. The breakpoint (in red) is located in the first intron. Part C shows the genomic sequence of the two breakpoint regions. Note the four nucleotides common to both chromosome 9 and chromosome 10 on der(10).
Since the initiation codon (ATG) of the KCNMA1 gene is located in the first exon, we hypothesized that the physical separation could cause an altered level of KCNMA1 transcript. We then performed a semiquantitative reverse transcription-polymerase chain reaction on total RNA isolated from lymphoblastoid cell lines of both the patient and a healthy comparison subject to verify that the translocation led to a lower expression of the KCNMA1 gene. Comparative analysis of the bands intensity with Bioprofil software (Vilber Lourmat, Marne La Vallee, France) between β-actin and KCNMA1 showed a KCNMA1 :β-actin ratio of 1.0 in the comparison subject and 0.55 in the patient (data not shown), suggesting the haploinsufficiency of the KCNMA1 gene.
Functional Studies
To verify that the reduced mRNA coding for KCNMA1 corresponded to similar reduced activity of the BK Ca channels, patch-clamp studies were performed in the patient’s lymphoblastoid cell lines ( Figure 2 ). Whole-cell currents were elicited in the presence of 400 nM Ca 2+ inside the cells by 8 mV depolarizing steps from −70 to +42 mV before and after blocking BK Ca channels with a specific blocker, Iberiotoxin. Whole cell recordings of K + currents were acquired in lymphoblastoid cell lines obtained from the autistic patient and the comparison subjects. While membrane capacitance was similar in both cell groups (16.3 pF [SD=0.7] and 17 pF [SD=0.9] in the comparison subjects and the patient, respectively), resting membrane potential and input membrane resistance were significantly different in the two cell groups (p<0.05). The changes were in agreement with reduced activity of a K + channel. Average resting membrane potential was –72.8 mV (SD=0.9) in the comparison subjects’ cells (N=12 cells) and –68.4 mV (SD=1.7) in the patient cells (N=12 cells), and average input resistance was 509 MΩ (SD=76) in the comparison subjects’ cells and 744 MΩ (SD=57) in the patient cells. The density of the IbTx-sensitive current in the patient’s cells was approximately one-half that of the comparison subjects’ cells, in accordance with reduced expression of mRNA coding for KCNMA1 ( Figure 2 ).
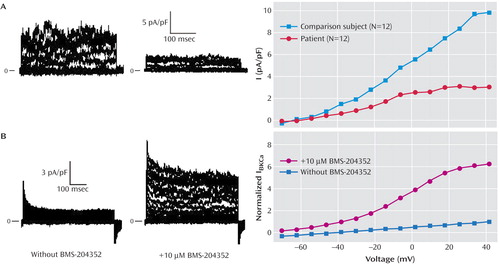
a Part A shows IbTx-sensitive currents in comparison subject and patient cells. Original traces of the difference currents before and after application of 100 nmol/liter IbTx on comparison subject cells (left) and patient cells (right). Currents were elicited by 8-mV depolarizing steps from –70 to +42 mV. Intracellular free Ca 2+ was set at 400 nM. Cell capacitance values were 14 pF (comparison subject cell) and 22 pF (patient cell). IbTx-sensitive currents on the averaged I-V relationship in comparison subject cells (N=12) and patient cells (N=12) are shown in the graph. Current density of patient cells was significantly lower relative to comparison subject values at the same voltage (p<0.05). Part B shows original traces of the difference currents before and after application of 10 μmol/liter BMS-204352 on a representative patient cell. The I-V relationship was obtained in similar conditions except for the presence of 4-AP to record mainly BK Ca current. In order to reveal mainly the effects of the drug, currents were normalized in each cell to the one elicited by the pulse to + 42 mV before the addition of BMS-204352; then the means and standard deviations were calculated on these normalized currents. Ten μmol/liter BMS-204352 significantly activated the current (N=7 cells) relative to control conditions (N=6 cells) (p<0.05).
These results suggested that one copy of the KCNMA1 gene was silenced in the autistic patient. Interestingly, the addition of the Ca 2+ -sensitive BK Ca opener, BMS-204532, a drug previously tested in patients with ischemic stroke (13) , significantly increased the activity of the remaining channels ( Figure 2 ) in the patient’s cells. To study the effects of BMS-204532, cells were first submitted to 4-AP to block most of the Kv channels found in the cells and then to BMS-204532 and 4-AP to observe the specific activation of the BK Ca channels.
Mutation Analysis
To verify that KCNMA1 is a possible candidate gene for autistic disorder, we investigated mutations in 116 Fragile X-negative patients with autism and a normal karyotype (46 Tunisian subjects, 40 Italian subjects, and 30 French subjects) by single-strand conformation polymorphism and denaturing high-performance liquid chromatography analyses. In one patient, we found one base-pair substitution in the second exon of KCNMA1 , changing an alanine to a valine, located in the first loop of the protein (part A of Figure 3 ). This sequence variation was not found in 225 Tunisian comparison subjects (450 chromosomes), and the confirmation of its absence in the other 115 subjects was verified by further denaturing high-performance liquid chromatography analyses. This intracellular region is highly conserved from C.elegans to mammals (part B of Figure 3 ), suggesting an important function. Furthermore, analysis of sequence motifs revealed that the corresponding heterozygous substitution might create a cryptic splice donor site in the second exon. The C-T exchange corresponded to position + 2 of a newly predicted splicing signal AGgt ggat that matched the consensus sequence almost perfectly, which was highly predicted (probability of 0.83) by the splice-prediction program Splice-View (http://125.itba.mi.cnr.it/~webgene/wwwspliceview.html). However, we were unable to test the functional consequences of the sequence variation in the KCNMA1 protein, since we had access to DNA only because of the intercurrent death (road accident) of the patient and his mother. Further functional analyses by site-directed mutagenesis would clarify the potential functional consequences of such a sequence variation.
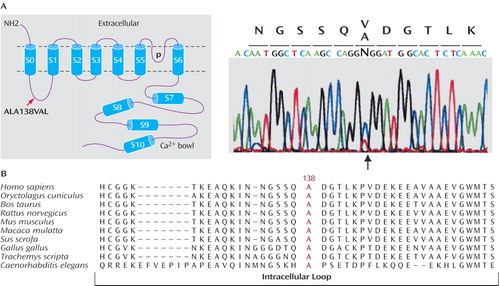
a Part A shows a schematic representation of the BK Ca channel structure and sequencing results of exon 2 in one Tunisian patient, showing the position of the ALA138VAL substitution. Part B shows the ClustalW sequence alignments of homologous BK Ca channels with the highly conserved alanine codon 138 (red) affected by the substitution.
Discussion
There have been a number of reports in the literature of chromosome aberrations in autism covering a broad spectrum of anomalies, including terminal and interstitial deletions, balanced and unbalanced translocations, and inversions (2 , 5) . However, particularly in the case of balanced translocations, none of these studies provide evidence of a functional defect associated with autism. In this study, we have presented arguments suggesting that the KCNMA1 gene is a strong candidate for autism and mental retardation.
Indeed, the KCNMA1 gene was physically disrupted on chromosome 10 by a balanced reciprocal translocation in a patient with autistic disorder and severe mental deficiency. The physical separation of the KCNMA1 gene promoter of the first exon from the remainder of the gene leads to a nonfunctional KCNMA1 allele (as shown in semiquantitative reverse transcription-polymerase chain reaction experiments). Although we found an associated 14 bp deletion at the breakpoints located in large noncoding regions, it is unlikely to have an effect on the decreased expression of the KCNMA1 transcript.
The 10q22 region has previously been suspected of being involved in autism and language disorders, with weak possible linkage to D10S201 (14) and D10S2327 (15) , located at 1.5 Mb and 1 Mb telomeric from KCNMA1 , respectively. The mapping of an autism susceptibility locus and a gene physically disrupted in the same chromosome region in an autistic patient suggest that this gene may be involved in this condition.
Large conductance calcium-activated potassium channels (BK Ca channels) are ubiquitously distributed among tissues, except in cardiac myocytes, and activated by membrane depolarization and increases of intracellular calcium concentration. They play a key role in the control of neuronal excitability, hormone secretion, and smooth muscle tone. Therefore, BK Ca channels are considered to be molecular integrators of biochemical and electrical signals.
The KCNMA1 gene is expressed abundantly in various human brain structures (e.g., amygdala, caudate nucleus, cerebral cortex, hippocampus, hypothalamus, spinal cord) (12) as well as in the cochlea (16) . In the vertebrate brain, the BK Ca channels are mainly located in the glutamatergic presynaptic active zone and might regulate synaptic transmission and transmitter release (17) . BK Ca channels are known to associate with β-subunit through the N -terminus, leading to specific regulation of channel function (18) . BK Ca channels have also been shown to associate with other proteins through the intracellular C-terminus region. In the brain, β2 adrenergic receptors form a macromolecular complex with BK Ca , specifically to regulate channel activity (19) . Other yeast two-hybrid screens have shown an interaction between BK Ca and 1) the light chain of the microtubule-associated protein 1A (MAP1A) which, with microtubules, is an important determinant of neuronal and nonneuronal cellular morphology (20) ; 2) β-catenin, which is involved in synapse organization and mediating cell adhesion (β-catenin associates with the synaptic protein Lin7/Velis/MALS, whose interaction partner, Lin2/CASK, also binds voltage-gated Ca 2+ channels; thus, β-catenin could provide a physical link between Ca 2+ and BK Ca channels at the presynaptic active zone) (21) ; and 3) syntaxin 1A, which is a soluble N -ethylmaleimide sensitive fusion attachment receptor protein required for the regulation of the activity and/or distribution of synaptic proteins within specialized cellular compartments (22) .
Mutations in the homologous D.melanogaster and C. elegans of the KCNMA1 gene (Slowpoke and Slo-1, respectively) have been shown to cause both electrophysiological defects and multiple behavioral abnormalities (23 , 24) . The recent report (25) on mice lacking BK Ca channels (-/-) showed that they presented cerebellar dysfunction and a dramatic reduction in the activity of the cerebellar Purkinje neurons. These data are very interesting, since many neuropathological observations on autistic individuals have revealed specific alterations of the Purkinje cells (26) .
Therefore, the finding in our study of a breakpoint in the KCNMA1 gene in the autistic patient with a de novo translocation, together with the predominant expression of KCNMA1 in the brain and during development, its chromosomal localization in a locus previously implicated in autism, and the finding of a sequence variation in an unrelated patient in a highly conserved region of the ion channel suggest that the KCNMA1 gene is a candidate for autistic disorder in humans. Furthermore, a recent report (27) described a Ca V 1.2 calcium channel dysfunction associated with autism, also suggesting the implication of Ca 2+ signaling in autism. Finally, Du and colleagues (28) recently reported a gain-of-function mutation in KCNMA1 associated with generalized epilepsy and paroxysmal movement disorder in a large family. Thus, alterations in neuronal excitability may contribute to different neurological disorders. Many potassium channel dysfunctions are associated with a range of neurological disorders, and drugs that target these channels might hold promise for clinical applications.
In summary, we report a channelopathy associated with autism and mental retardation, and we propose that a defect in BK Ca activity may abolish neuronal excitability. Future studies, including mutation analyses for KCNMA1 in large cohorts of autistic subjects, are needed as well as association studies with single nucleotide polymorphisms and screening for genomic rearrangements (i.e., deletions, duplications) located in the KCNMA1 genomic area. Finally, we aim to perform the functional study of the identified sequence variations (particularly the ALA138VAL substitution) with the reconstitution of the potentially mutant channel in appropriate heterologous cellular systems. This prospective work will evaluate a potential broader involvement of the KCNMA1 gene in autism and mental retardation and, more generally, help us to better understand the consequences of such synaptic defects associated with these psychiatric disorders.
1. Fombonne E: Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord 2003; 33:365–382Google Scholar
2. Folstein SE, Rosen-Sheidley B: Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet 2001; 2:943–955Google Scholar
3. Muhle R, Trentacoste SV, Rapin I: The genetics of autism. Pediatrics 2004; 113:e472–e486Google Scholar
4. Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, Nguyen L, Yang J, Harper C, Thorpe D, Vermeer S, Young H, Hebert J, Lin A, Ferguson J, Chiotti C, Wiese-Slater S, Rogers T, Salmon B, Nicholas P, Petersen PB, Pingree C, McMahon W, Wong DL, Cavalli-Sforza LL, Kraemer HC, Myers RM: A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 1999; 65:493–507Google Scholar
5. Castermans D, Wilquet V, Steyaert J, Van de Ven W, Fryns JP, Devriendt K: Chromosomal anomalies in individuals with autism: a strategy towards the identification of genes involved in autism. Autism 2004; 8:141–161Google Scholar
6. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T; Paris Autism Research International Sibpair Study: Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003; 34:27–29Google Scholar
7. Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthelemy C, Moraine C, Briault S: X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 2004; 74:552–557Google Scholar
8. Lord C, Rutter M, Le Couteur A: Autism Diagnostic Interview–Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 1994; 24:659–685Google Scholar
9. Schopler E, Reichler RJ, DeVellis RF, Daly K: Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J Autism Dev Disord 1980; 10:91–103Google Scholar
10. Schopler E, Reichler RJ, Bashford A, Lansing MD, Marcus LM: The Psychoeducational Profile Revised (PEP-R). Austin, Tex., Pro-Ed, 1990Google Scholar
11. Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML: Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem 1990; 265:11083–11090Google Scholar
12. Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH: Cloning, expression, and distribution of functionally distinct Ca(2+)-activated potassium channel isoforms from human brain. Neuron 1994; 13:1315–1330Google Scholar
13. Gribkoff VK, Starrett JE Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW: Targeting acute ischemic stoke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med 2001; 7:471–477Google Scholar
14. International Molecular Genetic Study of Autism Consortium (IGMSAC): A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet 2001; 69:570–581Google Scholar
15. Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH; Autism Genetic Research Exchange Consortium: Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet 2002; 70:60–71Google Scholar
16. Ramanathan K, Michael TH, Jiang JG, Hiel H, Fuchs PA: A molecular mechanism for electrical tuning of cochlear hair cells. Science 1999; 283:215–217Google Scholar
17. Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF: Presynaptic Ca 2+ -activated potassium channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci 2001; 21:9585–9597 Google Scholar
18. Orio P, Rojas P, Ferreira G, Latorre R: New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci 2002; 17:156–161Google Scholar
19. Liu G, Shi J, Yang L, Cao L, Park SM, Cui J, Marx SO: Assembly of a Ca 2+ -dependent BK channel signaling complex by binding to β2 adrenergic receptor. EMBO J 2004; 23:2196–2205 Google Scholar
20. Park SM, Liu G, Kubal A, Fury M, Cao L, Marx SO: Direct interaction between BK ca potassium channel and microtubule-associated protein 1A. FEBS Lett 2004; 570:143–148 Google Scholar
21. Lesage F, Hibino H, Hudspeth AJ: Association of beta-catenin with the alpha-subunit of neuronal large-conductance Ca 2+ -activated potassium channels. Proc Natl Acad Sci USA 2004; 101:671–675 Google Scholar
22. Ling S, Sheng JZ, Braun JE, Braun AP: Syntaxin 1A co-associates with native rat brain and cloned large conductance, calcium-activated potassium channels in situ. J Physiol 2003; 553:65–81Google Scholar
23. Atkinson NS, Brenner R, Chang W, Wilbur J, Larimer JL, Yu J: Molecular separation of two behavioral phenotypes by a mutation affecting the promoters of a Ca-activated K channel. J Neurosci 2000; 20:2988–2993Google Scholar
24. Wang ZW, Saifee O, Nonet ML, Salkoff L: SLO-1 potassium channels control quantat content of neurotransmitter release at the C elegans neuromuscular junction. Neuron 2001; 32:867–881 Google Scholar
25. Sausbier M, Hu H, Amtz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P: Cerebellar ataxia and Purkinje cell dysfunction caused by Ca 2+ -activated potassium channel deficiency. Proc Natl Acad Aci USA 2004; 101:9474–9478 Google Scholar
26. Palmen SJ, van Engeland H, Hof PR, Schmitz C: Neuropathological findings in autism. Brain 2004; 127:2572–2583Google Scholar
27. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT: Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119:19–31Google Scholar
28. Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, L Ders HO, Shi J, Cui J, Richerson GB, Wang QK: Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 2005; 37:733–738Google Scholar

