
5-HT2 and D2 Receptor Occupancy of Olanzapine in Schizophrenia: A PET Investigation
Abstract
OBJECTIVE: Olanzapine is a new atypical antipsychotic recently introduced for the treatment of schizophrenia. The purpose of this study was to investigate olanzapine's binding to the serotonin 5-HT2 and dopamine D2 receptors in schizophrenic patients being treated with clinically relevant doses. METHOD: Twelve patients with schizophrenia were randomly assigned to 5, 10, 15, or 20 mg/day of olanzapine in a prospective fashion. Three other subjects taking 30–40 mg/day were also included. Once steady-state plasma levels were achieved, dopamine D2 and serotonin 5-HT2 receptors were assessed by using [11C]raclopride and [18F]setoperone positron emission tomography imaging, respectively. Ratings of clinical status, extrapyramidal side effects, and prolactin levels were also obtained. RESULTS: Olanzapine induced near saturation of the 5-HT2 receptors, even at 5 mg/day. Its D2 occupancy increased with dose: patients taking 5–20 mg/day showed 43%–80% D2 occupancy, while patients taking 30–40 mg/day showed 83%–88%. CONCLUSIONS: Olanzapine is a potent 5-HT2 blocker and shows a higher 5-HT2 than D2 occupancy at all doses. However, its D2 occupancy is higher than that of clozapine and similar to that of risperidone. In the usual clinical dose range of 10–20 mg/day, its occupancy varies from 71% to 80%, and this restricted range may explain its freedom from extrapyramidal side effects and prolactin elevation. However, doses of 30 mg/day and higher are associated with more than 80% D2 occupancy and may have a higher likelihood of prolactin elevation and extrapyramidal side effects. (Am J Psychiatry 1998; 155:921–928)
For the past three decades the mainstay of pharmacological treatment of schizophrenia was the “typical” neuroleptics, which were predominantly dopamine D2 receptor blockers. These drugs were effective in controlling positive symptoms but had only a partial effect on the negative symptoms and cognitive dysfunction of schizophrenia. Furthermore, because of their prominent blockade of the dopamine D2 receptors, these drugs are associated with a high prevalence of acute extrapyramidal side effects as well as tardive dyskinesia, and they elevate prolactin levels, leading to sexual and endocrine side effects. The introduction of clozapine radically changed thinking about the mechanism of action of antipsychotics. Clozapine is a weak blocker of the dopamine D2 receptor and shows significantly more affinity for a range of receptors, especially the serotonin 5-HT2 receptor. At a clinical level, clozapine shows a superior efficacy on negative symptoms and is practically free of extrapyramidal side effects, tardive dyskinesia, and prolactin elevation. Thus, clozapine has served as a template for the development of the next generation of “atypical” antipsychotics (e.g., risperidone, olanzapine, sertindole, and quetiapine). While the precise mechanism of the atypicality of clozapine is unclear, one prominent hypothesis implicates its high affinity for the 5-HT2 receptor, combined with its low affinity for the dopamine D2 receptor (1–4).
Positron emission tomography (PET) neuroreceptor studies have made it possible to contrast the pharmacological effects of typical and atypical drugs in patients. In routine clinical doses, typical neuroleptics occupy between 70% and 90% of the striatal dopamine D2 receptors (5). Less than 60% D2 blockade may not be sufficient to induce a satisfactory antipsychotic response with typical neuroleptics (6), while D2 occupancies of greater than 80% are associated with a higher prevalence of extrapyramidal side effects (5, 6). The fore~going facts suggest that when used in doses that lead to between 60% and 80% dopamine D2 receptor blockade, typical antipsychotics may induce antipsychotic response with few extrapyramidal side effects (7).
The situation is different with clozapine, the prototypical atypical antipsychotic. First, clozapine occupies between 24% and 66% of the dopamine D2 receptors at most clinical doses (75–900 mg/day) (8, 9). Second, it demonstrates a higher occupancy of the 5-HT2 than the D2 receptor, consistent with its high in vitro affinity for the 5-HT2 receptors (8, 9). These data are of particular interest since they demonstrate that clozapine is an effective antipsychotic at levels of dopamine D2 receptor blockade that in all likelihood would not be sufficient, in and of themselves, to induce an antipsychotic response.
It is of interest, then, to evaluate whether the newer agents replicate these pharmacological properties of clozapine. Risperidone shares with clozapine a high (greater than 80%) occupancy of 5-HT2 receptors, but unlike clozapine it is efficacious only at doses that induce 66%–80% D2 occupancy (i.e., 2–6 mg/day) (10). Furthermore, those doses of risperidone that show greater than 80% D2 occupancy (i.e., doses above 6 mg/day) do not show a consistent superiority over haloperidol with respect to extrapyramidal side effects; and their beneficial effect on negative symptoms is also diminished (11).
Olanzapine, another newly marketed atypical antipsychotic, is chemically similar to clozapine and shares several aspects of clozapine's in vitro pharmacological profile (stronger affinities for the 5-HT2, muscarinic, and histaminic receptors than for the dopamine D2 receptor) (12). These pharmacological characteristics translate into clozapine-like clinical benefits: substantially reduced extrapyramidal side effects, less effect on prolactin, and probably a direct effect on ameliorating negative symptoms (13–16). These clinical studies suggest that the optimal dose for olanzapine may be between 10 and 20 mg/day. Therefore, it is of clinical and scientific interest to determine olanzapine's in vivo receptor occupancy profile in patients at clinically relevant doses. In particular, we asked 1) Does olanzapine (like clozapine and risperidone) have a potent effect on 5-HT2 receptors? 2) What is olanzapine's D2 occupancy in its effective dose range? 3) Does olanzapine (like clozapine and risperidone) have a greater effect on the 5-HT2 as opposed to the D2 receptors? 4) At what doses, if any, does olanzapine demonstrate greater than 80% D2 occupancy? 5) If its D2 occupancy does go beyond 80%, what implication does this have for prolactin levels and extrapyramidal side effects?
Previous neuroreceptor occupancy studies of antipsychotics have had two prominent methodological limitations. Studies using normal subjects have usually studied the drug effect after a single dose (17, 18). This design does not reflect the clinical situation that always entails multiple dosing to steady state. On the other hand, studies of patients have mostly entailed the use of extended case series of patients who were being treated clinically without random assignment to dose and without control over dose titration (5, 8, 10). To overcome these confounds and to obtain a valid estimate of olanzapine's effect on the dopamine D2 and serotonin 5-HT2 receptors, we studied patients with schizophrenia, who were randomly assigned, in a prospective fashion, to take a fixed-multiple-dose regimen of a clinically relevant dose of olanzapine.
METHOD
This study was approved by the Human Subjects Review Committee of the University of Toronto. Patients participated after receiving detailed information about the study and signing a written consent document. Male and female patients were included if they were between the ages of 18 and 45 years and met DSM-IV criteria for a diagnosis of schizophrenia. Patients were excluded from participation if they suffered from a major medical or neurological illness, if they met DSM-IV criteria for substance abuse in the last 3 months, if they had received a depot antipsychotic medication in the last 6 months, or if they were receiving a concomitant psychotropic medication (benzodiazepine and antiparkinsonian agents were administered).
The study was designed to obtain PET data on three subjects in each of the four dose groups, 5 mg/day, 10 mg/day, 15 mg/day, and 20 mg/day, by random assignment. Three subjects dropped out before the PET scan and were replaced by random assignment. In addition to these data, three scans were obtained for three patients receiving treatment with 30, 30, and 40 mg/day of olanzapine, respectively. Two of these scans were for individuals who had participated in the study and did not show a satisfactory response at their assigned dose, and the third scan was for an individual receiving clinical treatment with 30 mg/day.
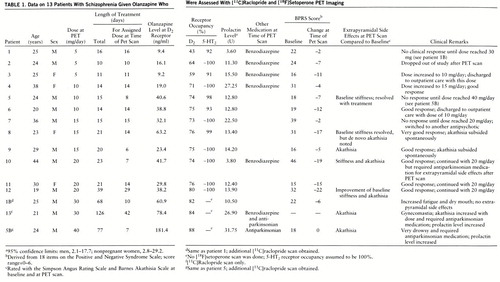
Patients enrolled in the study went through a washout period from their previous neuroleptic that lasted 2–4 days, and medication was then titrated to their assigned dose. Patients assigned to 5 and 10 mg/day started with these doses. Those assigned to 15 and 20 mg/day were started at 10 mg/day, and their dose was increased in weekly increments of 5 mg/day. Patients were held at their assigned target dose until steady-state levels were achieved (5 days or greater), at which stage PET scans were done. Two of the patients were neuroleptic naive, another two had not had any neuroleptic for more than 6 months, and the rest had at least a 14-day washout from their previous neuroleptic at the time of PET scanning during olanzapine treatment. The D2 occupancy was assessed by using [11C]raclopride, 12 hours after the last dose, while the 5-HT2 occupancy was assessed on the same day 2–3 hours after the D2 PET scan (14–15 hours after the last dose). After the PET scan the patients reverted to a flexible-dosing, open clinical treatment and were followed for a maximum of 8 weeks. To determine their clinical status, each of the patients was rated with the Positive and Negative Syndrome Scale (19) and a Positive and Negative Syndrome Scale-derived Brief Psychiatric Rating Scale (20) at baseline, at the time of the PET scan, and at end of their participation in the open clinical phase. At these times, extrapyramidal side effects were assessed with the Barnes Akathisia Scale (21) and the Simpson Angus Rating Scale (22).
The PET scans to estimate D2 occupancy were obtained after the injection of 10 mCi of high-specific-activity [11C]raclopride (300–1600 Ci/mmol), through use of a bolus plus infusion protocol. The methods employed here are identical to those described in previous reports in this journal (7). The pertinent aspects are the following: striatal and cerebellar regions of interest were drawn on two contiguous PET slices on a composite PET image with reference to an MRI scan (General Electric Signa 1.5-T scanner, T2-weighted spin-echo sequence) coregistered to the PET scan by using a surface-matching algorithm as implemented in ANALYZE 7.0 (Biomedical Imaging Resource, Rochester, Minn.). An estimate of binding potential of raclopride (D2BP) (which represents the Bmax/Kd of [11C]~raclopride for D2 receptors; where Bmax is the total number of receptors available to a ligand and Kd the affinity of the ligand for the receptors) was obtained from the ratio of the striatum to the cerebellum. As used in our laboratory, this method yields a test-retest standard deviation of 6% and has been standardized to have a high interrater and intrarater reliability (intraclass correlation coefficient [ICC] greater than 0.95).
To estimate receptor occupancy, we used an age-corrected baseline derived from a pool of 16 normal control subjects and 12 neuroleptic-naive patients with schizophrenia. Since the illness has no statistically discernible effect on D2 receptors as measured with [11C]raclopride (23), the data from neuroleptic-naive patients and control subjects were pooled to get a better estimate of the effect of age on D2BP (F=0.66, df=1,25, p=0.42). Olanzapine-induced D2 receptor occupancy was determined as (D2BPBas–D2BPOlz)/(D2BPBas), where D2BPBas is the age-corrected D2BP baseline and D2BPOlz is the D2BP for patients on a regimen of olanzapine. The absence of patients' own baseline values introduces a potential error; the error, as calculated on the basis of variance in data from neuroleptic-naive patients, is expected to vary from 0% to 9% for patients with 50% occupancy and 0% to 4% for patients who have 80% occupancy (24).
The 5-HT2 scans were obtained by using a bolus injection of 5 mCi of high-specific-activity [18F]setoperone (360–6210 Ci/mmol), after the method developed and standardized by Blin et al. (25, 26). The 5-HT2 occupancy was determined in the prefrontal cortex regions of interest drawn on the [18F]setoperone scan with reference to a coregistered MRI (as described earlier). An index of the 5-HT2 receptors was obtained from the prefrontal cortex to cerebellum ratio over the 65–90-minute time period. The cerebellum is practically devoid of 5-HT2 receptors (27), and studies of baboons as well as of humans report no displaceable [18F]setoperone binding in this region (25, 26, 28). It can be shown that at a time when the binding of the radioligand is at pseudoequilibrium, the prefrontal to cerebellum ratio represents binding potential + 1 (29). The details of this method as applied here have been described elsewhere (30). This method yields an average test-retest deviation of 6%–7% and an acceptably high interrater reliability (ICC r>>0.95) (30).
Since these patients were already receiving treatment, it was not possible to measure their baseline 5-HT2 binding potential. In the absence of this baseline, we used the age-corrected 5-HT2 binding potential obtained from 11 neuroleptic-free patients with schizophrenia and 26 age-matched normal control subjects. The pooling of normal control subjects and patients results in a more robust age-corrected regression and is justified, since there was neither an effect of diagnosis (F=1.23, df=1,33, p=0.28) nor a significant effect of diagnosis on the age regression (F=0.59, df=1,33, p=0.45). Occupancy was calculated in the same way as for dopamine D2 occupancy.
Coincident with the PET scans, blood was drawn for olanzapine and prolactin analysis. The levels of olanzapine were estimated in heparinized human plasma by using high performance liquid chromatography with electrochemical detection (BAS Analytics, West Lafayette, Ind.). Prolactin levels were determined by using a two-site cheminoluminometric immunoassay with a minimum detectable limit of 0.3 ng/ml and a coefficient of variance of 3.6%–4.5% (ACS, CIBA-Corning Diagnostics). Statistical analysis was implemented by using STATISTICA release 5 (StatSoft, Tulsa, Okla.).
RESULTS
Twelve patients (patients 1–12 in table 1) each had a [11C]raclopride and a [18F]setoperone scan, and three additional [11C]raclopride scans (for patients 1B, 5B, and 13) were obtained; data on all the scans are reported. Plasma levels at the time of the [11C]raclopride PET scan, 12 hours after the last dose, were linearly related to dose as plasma level=5.8 + 1.99×dose (F=22.16, df=1,12, multiple R=0.80, p≤0.001). The plasma levels at the time of the setoperone scan (14–15 hours after the last dose) were slightly lower than those at the time of the raclopride scan (on average, 8% lower) (paired t test t=2.63, df=11, p<0.05) but, understand~ably, were highly correlated (F=273.38, df=1,10, multiple R=0.98, p<0.001).
Olanzapine induced near saturation of the 5-HT2 receptors at all doses used in this study. All patients showed greater than 90% 5-HT2 occupancy, even at 5 mg/day.
The D2 occupancy varied as a function of dose. It was an average of 55% with 5 mg/day, 73% with 10 mg/day, 75% with 15 mg/day, and 76% with 20 mg/day. The average with 30 mg/day was 83%, and the single patient on a regimen of 40 mg/day showed 88%. The expected relationship between dose/plasma levels and D2 occupancy is that of a saturating rectangular hyperbola defined by the following equation: percent occupancy=100 (dose/(dose + ED50), where ED50 is the dose/plasma level that occupies 50% of the available receptors. This function fits the data quite well with an ED50 of 4.5 mg/day of olanzapine in terms of oral dose (N=15, R=0.84, 70% of variance explained) and with an ED50 of 10.3 ng/ml in terms of olanzapine plasma levels (N=15, R=0.83, 69% of variance explained) (figure 1).
Patients were scanned at variable intervals after the start of olanzapine. This aspect of the design, along with the small group size, makes it difficult to draw reliable conclusions regarding the relationship between dose/plasma levels and clinical response in these data. Therefore, we have chosen to present the clinical response data on each of the patients in table 1. In terms of side effects, three patients (patients 5, 8, and 12) in the 10–20-mg/day range noted a resolution of their baseline extrapyramidal side effects, which in all likelihood reflected a resolution of the carryover effects of the previous antipsychotic at the time of the baseline rating. On the other hand, two (patients 9 and 10) developed motor side effects with olanzapine; one developed akathisia that subsided spontaneously, while the other developed akathisia and stiffness that required antiparkinsonian medications.
Of the patients who did not respond satisfactorily to their assigned dose, two (patients 1 and 5) had their dose increased systematically to 30 and 40 mg/day but without further benefit. These patients had their D2 occupancies estimated at the higher doses and showed the expected increase in receptor occupancy (patient 1's dose was increased from 5 mg/day to 30 mg/day, and occupancy increased from 43% to 82%; patient 5's dose was increased from 10 mg/day to 40 mg/day, and occupancy increased from 74% to 88%). The second scan confirms that the lack of response at the higher dose was not due to lack of sufficient D2 occupancy. The patient who was in routine clinical treatment at 30 mg/day showed 84% D2 occupancy. Of the three patients with doses above 20 mg/day, two experienced akathisia (one with 30 mg/day and one with 40 mg/day) requiring antiparkinsonian medications.
Prolactin levels at the time of PET scans were also obtained. Of the patients in the 5–20-mg/day range, only one showed prolactin levels above the normal range (table 1). Of the three patients with doses above 20 mg/day, two showed abnormal elevations. The subject (patient 5) whose dose was titrated from 10 to 40 mg/day showed a prolactin increase from 12.8 to 31.8 ng/ml (normal range=2.1–17.7 ng/ml), while the subject (patient 13) scanned once while taking 30 mg/day showed a value of 26.9 ng/ml. The third subject with a dose above 20 mg/day (patient 1) also showed an increase in prolactin level, from 3.6 to 10.5 ng/ml as his dose was increased from 5 to 30 mg/day. These data are to be interpreted with caution, since we do not have the subjects' own baseline (i.e., pre-olanzapine) estimates. Even if we had obtained baseline prolactin levels, the levels would be confounded because a 2–4-day washout does not eliminate the carryover effects of the previous antipsychotic. Furthermore, it has been shown previously that if prolactin elevations are noted at all at doses of 10–20 mg/day, they are generally transient. Because we determined prolactin levels only at a single point in time, we are unable to determine the persistence or transience of the levels observed at 30 and 40 mg/day. Nonetheless, until better data are available, it is valuable to consider that these data suggest that doses above 20 mg/day may be associated with a higher likelihood of prolactin elevation.
DISCUSSION
Olanzapine shows a combination of atypical and typical characteristics, as determined with PET imaging. Like clozapine, olanzapine has a potent effect on the 5-HT2 receptors. Even the lowest clinical dose of 5 mg/day occupies greater than 90% of 5-HT2 receptors. On the other hand, unlike clozapine and like risperidone, olanzapine, in routine clinical doses, occupies greater than 60% of D2 receptors and, if used at doses above 20 mg/day, occupies greater than 80% of dopamine D2 receptors. At doses above 20 mg/day, which led to greater than 80% D2 occupancy, extrapyramidal side effects and prolactin elevation were also noted. We discuss the implications of these findings for clinical practice as well as for understanding the mechanism of antipsychotic action.
This study constitutes the first systematic analysis of the in vivo occupancy of olanzapine and extends and clarifies two previous reports. Pilowsky and colleagues (31) have reported that the D2 occupancy of olanzapine, through use of I123-Iodobenzamide (IBZM) single photon emission computed tomography (SPECT) imaging, is similar to that of clozapine and lower than that of typical neuroleptics. However, the report of Pilowsky et al. has several limitations. The spatial resolution, quantification, and signal/noise characteristics of IBZM-SPECT imaging are inferior to those of [11C]~raclopride PET imaging. The study had only six patients (four on a regimen of 10 mg/day, one each on a regimen of 15 and 20 mg/day), nonrandom dose assignment, flexible dose titration, and variable duration before scanning. It did not control for age-related D2 changes, and plasma levels were not available to confirm compliance. Furthermore, the study found no relationship between dose and occupancy, questioning the internal consistency of the data. The present study overcomes these limitations. The other previous data come from Nyberg et al. (18), who studied three normal control subjects after a single dose of 10 mg of olanzapine. Seven hours after the dose the subjects had olanzapine plasma levels of 10–12 ng/ml, with 59%–63% D2 occupancy. Nyberg et al. predicted that patients taking 10 mg/day would have 75% occupancy at steady state—very close to the 73% (SD=2%) that we observed. Given the limitations of the Pilowsky et al. data, and the concordance of our results with the Nyberg et al. prediction, we feel confident that our conclusion that olanzapine D2 occupancy is higher than that of clozapine and comparable to that of typical neuroleptics is correct.
Meltzer et al. (1, 32) have suggested that the distinguishing pharmacological characteristic of an atypical antipsychotic is a higher affinity for 5-HT2 than D2 receptors in vitro (1, 32). In patients this characteristic manifests itself as greater than 80% 5-HT2 occupancy along with less than 80% D2 occupancy (3, 4). By these accounts, olanzapine's in vivo pharmacology is clearly atypical. It is to be noted that olanzapine differs from typical neuroleptics along several other dimensions. In particular, it has prominent actions on the dopamine D4 and D1, muscarinic, and histaminic receptors (12), any of which may, in principle, contribute to its clinical atypicality. However, at this time there are no studies of olanzapine's actions at these other receptors in humans.
While other receptors may be of additive interest in the newer atypical antipsychotics, the role of D2 blockade is still central. Risperidone, for example, is proven to be effective only at doses that block greater than 60% of D2 receptors (i.e., a dose of 2 mg/day), and doses that lead to greater than 80% dopamine D2 blockade (i.e., doses above 6 mg/day) tend to lose some of their atypical characteristics (10, 11, 33). Olanzapine also seems to share this characteristic. It has been proven to be superior to placebo in the 10–20-mg/day range (13, 14, 16), which, as we observe here, leads to 70%–80% D2 occupancy. Our data show that doses greater than 20 mg/day lead to higher than 80% occupancy; and this was associated with extrapyramidal side effects as well as prolactin elevation. Because of its built-in anticholinergic activity, it is likely that patients may be able to tolerate doses higher than 20 mg/day without manifest extrapyramidal side effects (akin to using haloperidol with greater than 80% D2 occupancy but under cover of an anticholinergic). A recent report (uncontrolled case series) on the use of high-dose olanzapine (30–40 mg/day) in refractory patients found that five of eight patients experienced motor side effects, three with akathisia and four with parkinsonism, although the symptoms were mild for most patients and required medication in only one (34). Therefore, until more systematic clinical trials are done at doses higher than 20 mg/day, it should not be assumed that the relative freedom from extrapyramidal side effects seen in the 10–20-mg/day range (13, 14, 16) will apply to doses beyond 20 mg/day. On the basis of our PET findings, it is quite likely that doses of olanzapine higher than 20 mg/day will not exhibit the virtually complete freedom from extrapyramidal side effects and prolactin elevation that is observed in the 10–20-mg/day range.
These findings raise interesting questions regarding the comparison of olanzapine to typical neuroleptics and to clozapine. All currently published comparisons of olanzapine have used 10–20 mg/day of haloperidol as a reference drug. Thus, doses of olanzapine that result in 70% to 80% D2 occupancy have been compared to doses of haloperidol that would result in greater than 90% D2 occupancy for most patients (35). It is increasingly realized that the optimal dose of haloperidol may be lower than 10 mg/day and that higher doses may result in greater extrapyramidal side effects and in higher negative symptom ratings (36–38). Furthermore, it has also been shown that doses which induce occupancies higher than 80% with typical neuroleptics are associated with a greater prevalence of extrapyramidal side effects (5, 6). These facts raise an interesting question: would olanzapine demonstrate benefits on negative symptoms, extrapyramidal side effects, and prolactin that are superior to those of a typical neuroleptic, if their D2 occupancies were matched one to one?
Olanzapine's D2 occupancy is also of interest when compared with that of clozapine. Olanzapine is chemically very similar to clozapine and in vitro seems to have the same relative receptor affinity profile. However, its absolute affinity for the dopamine D2 receptor is twenty-five to fifty times higher than that of clozapine. Therefore, the comparison of its D2 occupancy in a clinical situation is of interest. While clozapine occupies, on average, 30%–60% of D2 receptors at therapeutic doses (8, 9), olanzapine occupies 70%–80%. This is an important distinction. It has been shown that greater than 60% dopamine D2 occupancy may be sufficient to induce antipsychotic response in and of itself, while less than 60% dopamine D2 occupancy may be insufficient (6). Thus, while clozapine does not call on the typical D2 mechanism for inducing clinical response, olanzapine in all likelihood does.
While the ultimate clinical significance of this difference between clozapine and olanzapine can be determined only in a direct clinical comparison, the D2 data obtained here provide grounds for hypothesizing a difference. Clinical data indicate that all atypical antipsychotics are not alike. For example, risperidone is not particularly effective for individuals who have not responded to clozapine, whereas clozapine can be beneficial in up to 50% of individuals who have not responded to risperidone (39–41). A direct comparison between olanzapine and clozapine, and data on the use of each in patients who have not responded to the other, would provide the clinical data to understand whether this difference in D2 occupancy manifests itself clinically.
These data need to be interpreted in light of the salient limitations of this study. The group size of the study was chosen to address the primary objective, i.e., the in vivo profile of olanzapine with respect to 5-HT2 and D2 occupancy at clinical doses in patients. This group is much smaller than would be required to definitively analyze the relationship between dose/receptor occupancy and clinical response. Since the dose-response relationships of olanzapine are well established from previous multicenter, fixed-dose clinical trials (13, 14, 16), patients in this study were randomly assigned to the same doses in fixed-dose regimens so that the PET data could be related to the previously published clinical data. A second limitation of this study is the measure of D2 occupancy. [11C]Raclopride is the most reliable and standardized ligand for the determination of D2 occupancy; however, it provides data only for the striatum. The exact site of antipsychotic response is not known, but it is speculated that mesolimbic dopamine D2 receptors may be crucial determinants. Because of a low density of D2 receptors in the mesolimbic regions, they are not reliably visualized with [11C]raclopride. Since it has been demonstrated that olanzapine shows the same affinity for the striatal and mesolimbic D2 receptors (42), it is fair to consider striatal D2 occupancy as a surrogate for mesolimbic D2 occupancy, until more direct measures of mesolimbic occupancy are available (43).
In summary, olanzapine saturates 5-HT2 receptors and demonstrates a higher 5-HT2 than D2 occupancy at all doses. However, its D2 occupancy is higher than that of clozapine and comparable to that reported previously for typical neuroleptics and risperidone. Thus, at doses that give higher D2 occupancies (particularly at doses greater than 20 mg/day that give higher than 80% occupancy), it may show a higher prevalence of extrapyramidal side effects and prolactin elevation. Thus, while olanzapine is a well-tolerated atypical antipsychotic in the 10–20-mg/day dose range, it may lose some of its atypical clinical benefits if used at higher doses.
 |
Received Oct. 20, 1997; revision received Feb. 6, 1998; accepted Feb. 16, 1998. From the Schizophrenia Division and PET Centre, The Clarke Institute of Psychiatry, Department of Psychiatry, University of Toronto. Address reprint requests to Dr. Kapur, PET Centre, The Clarke Institute of Psychiatry, 250 College St., Toronto, Ont., Canada M5T 1R8; [email protected] (e-mail). Supported by an award from the Medical Research Council of Canada (Dr. Kapur) and by Eli Lilly of Canada.Astra Arcus AB provided the precursor used in the synthesis of [11C]raclopride and Janssen-Cilag, the precursor for the radiochemical synthesis of [18F]setoperone. The authors thank Erin Toole, Doug Hussey, Kevin Cheung, and Terry Bell for their technical assistance.

FIGURE 1. Relationship Between Dopamine D2 and Serotonin 5-HT2 Receptor Occupancy and Olanzapine Dose and Plasma Levela
aThe curve represents a saturating hyperbola that best describes the relationship between D2 occupancy and both dose and plasma level.
1 Meltzer HY, Matsubara S, Lee JC: The ratios of serotonin-2 and dopamine-2 affinities differentiate atypical and typical antipsychotic drugs. Psychopharmacol Bull 1989; 25:390–392Medline, Google Scholar
2 Meltzer HY: The role of serotonin in schizophrenia and the place of serotonin-dopamine antagonist antipsychotics. J Clin Psychopharmacol 1995; 15:2S–3SCrossref, Medline, Google Scholar
3 Kapur S, Remington G: Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry 1996; 153:466–476Link, Google Scholar
4 Kapur S: A new framework for investigating antipsychotic action in humans: Lessons from PET Imaging. Molecular Psychiatry 1998 (in press)Google Scholar
5 Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G: Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine: relation to extrapyramidal side effects. Arch Gen Psychiatry 1992; 49:538–544Crossref, Medline, Google Scholar
6 Nordstrom AL, Farde L, Wiesel FA, Forslund K, Pauli S, Halldin C, Uppfeldt G: Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects—a double-blind PET study of schizophrenic patients. Biol Psychiatry 1993; 33:227–235Crossref, Medline, Google Scholar
7 Kapur S, Remington G, Jones C, Wilson A, DaSilva J, Houle S, Zipursky R: High levels of dopamine D2 receptor occupancy with low-dose haloperidol treatment: a PET study. Am J Psychiatry 1996; 153:948–950Link, Google Scholar
8 Nordström A-L, Farde L, Nyberg S, Karlsson P, Halldin C, Sedvall G: D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry 1995; 152:1444–1449Link, Google Scholar
9 Zipursky R, Kapur S, Jones C, Remington G, Shammi C, Houle S: Is there a “glass ceiling” to the D2 occupancy of clozapine? (abstract). J Psychopharmacol 1997; 11:A73Google Scholar
10 Kapur S, Remington G, Zipursky RB, Wilson AA, Houle S: The D2 dopamine receptor occupancy of risperidone and its relationship to extrapyramidal symptoms: a PET study. Life Sci 1995; 57:PL103–PL107Google Scholar
11 Chouinard G, Jones B, Remington G, Bloom D, Addington D, MacEwan GW, Labelle A, Beauclair L, Arnott W: A Canadian multicenter placebo-controlled study of fixed doses of risperidone and haloperidol in the treatment of chronic schizophrenic patients. J Clin Psychopharmacol 1993; 13:25–40Crossref, Medline, Google Scholar
12 Bymaster FP, Calligaro DO, Falcone JF, Marsh RD, Moore NA, Tye NC, Seeman P, Wong DT: Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacol~ogy 1996; 14:87–96Crossref, Medline, Google Scholar
13 Beasley CM Jr, Sanger T, Satterlee W, Tollefson G, Tran P, Hamilton S: Olanzapine versus placebo: results of a double-blind, fixed-dose olanzapine trial. Psychopharmacology (Berl) 1996; 124:159–167Crossref, Medline, Google Scholar
14 Beasley CM Jr, Tollefson G, Tran P, Satterlee W, Sanger T, Hamilton S: Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology 1996; 14:111–123Crossref, Medline, Google Scholar
15 Tollefson GD, Sanger TM: Negative symptoms: a path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am J Psychiatry 1997; 154:466–474Link, Google Scholar
16 Tollefson GD, Beasley CM Jr, Tran PV, Street JS, Krueger JA, Tamura RN, Graffeo KA, Thieme ME: Olanzapine versus haloperidol in the treatment of schizophrenia and schizoaffective and schizophreniform disorders: results of an international collaborative trial. Am J Psychiatry 1997; 154:457–465Link, Google Scholar
17 Nyberg S, Dahl ML, Halldin C: A PET study of D2 and 5-HT2 receptor occupancy induced by risperidone in poor metabolizers of debrisoquin and risperidone. Psychopharmacology (Berl) 1995; 119:345–348Crossref, Medline, Google Scholar
18 Nyberg S, Farde L, Halldin C: A PET study of 5-HT2 and D2 dopamine receptor occupancy induced by olanzapine in healthy subjects. Neuropsychopharmacology 1997; 16:1–7Crossref, Medline, Google Scholar
19 Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276Crossref, Medline, Google Scholar
20 Overall JE, Gorham DR: The Brief Psychiatric Rating Scale, in Handbook of Psychiatric Rating Scales, 2nd ed. By Lyerly SB. Rockville, Md, National Institute of Mental Health, Clinical Research Branch, 1973, pp 23–25Google Scholar
21 Barnes TRE: A rating scale for drug-induced akathisia. Br J Psychiatry 1989; 154:672–676Crossref, Medline, Google Scholar
22 Simpson GM, Angus JWS: A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 1970; 212:11–19Crossref, Medline, Google Scholar
23 Farde L, Wiesel FA, Stone-Elander S, Halldin C, Nordstrom AL, Hall H, Sedvall G: D2 dopamine receptors in neuroleptic-naive schizophrenic patients. Arch Gen Psychiatry 1990; 47:213–219Crossref, Medline, Google Scholar
24 Kapur S, Zipursky R, Jones C, Remington G, Wilson A, DaSilva J, Houle S: The D2 receptor occupancy profile of loxapine determined using PET. Neuropsychopharmacology 1996; 15:562–566Crossref, Medline, Google Scholar
25 Blin J, Pappata S, Kiyosawa M, Crouzel C, Baron JC: [18F]Setoperone: a new high-affinity ligand for positron emission tomography study of the serotonin-2 receptors in baboon brain in vivo. Eur J Pharmacol 1988; 147:73–82Crossref, Medline, Google Scholar
26 Blin J, Sette G, Fiorelli M, Bletry O, Elghozi JL, Crouzel C, Baron JC: A method for the in vivo investigation of the serotonergic 5-HT2 receptors in the human cerebral cortex using positron emission tomography and 18F-labeled setoperone. J Neurochem 1990; 54:1744–1754Crossref, Medline, Google Scholar
27 Pazos A, Probst A, Palacios JM: Serotonin receptors in the human brain, IV: autoradiographic mapping of serotonin-2 receptors. Neuroscience 1987; 21:123–139Crossref, Medline, Google Scholar
28 Petit-Taboue MC, Landeau B, Osmont A, Tillet I, Barre L, Baron JC: Estimation of neocortical serotonin-2 receptor binding potential by single-dose fluorine-18-setoperone kinetic PET data analysis. J Nucl Med 1996; 37:95–104Medline, Google Scholar
29 Mazoyer B: Investigation of the dopamine system with positron emission tomography: general issues in modelling, in Brain Dopaminergic Imaging With Positron Tomography. Edited by Baron JC. Dordrecht, The Netherlands, Kluwer Academic, 1991, pp 64–83Google Scholar
30 Kapur S, Jones C, DaSilva J, Wilson A, Houle S: Reliability of a simple non-invasive method for the evaluation of 5-HT2 receptors using [18F]-setoperone PET imaging. Nucl Med Commun 1997; 18:395–399Crossref, Medline, Google Scholar
31 Pilowsky LS, Busatto GF, Taylor M, Costa DC, Sharma T, Sigmundsson T, Ell PJ, Nohria V, Kerwin RW: Dopamine D-2 receptor occupancy in vivo by the novel atypical antipsychotic olanzapine—a I-123 IBZM single photon emission tomography (SPET) study. Psychopharmacology (Berl) 1996; 124:148–153Crossref, Medline, Google Scholar
32 Meltzer HY, Matsubara S, Lee JC: Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin-2 pKi values. J Pharmacol Exp Ther 1989; 251:238–246Medline, Google Scholar
33 Farde L, Nyberg S, Oxenstierna G, Nakashima Y, Halldin C, Ericsson B: Positron emission tomography studies on D-2 and 5-HT2 receptor binding in risperidone-treated schizophrenic patients. J Clin Psychopharmacol 1995; 15:S19–S23Google Scholar
34 Sheitman BB, Lindgren JC, Early J, Sved M: High-dose olanzapine for treatment-refractory schizophrenia (letter). Am J Psychiatry 1997; 154:1626Link, Google Scholar
35 Kapur S, Zipursky R, Roy P, Jones C, Remington G, Reed K, Houle S: The relationship between D2 receptor occupancy and plasma levels on low dose oral haloperidol: a PET study. Psychopharmacology (Berl) 1997; 131:148–152Crossref, Medline, Google Scholar
36 McEvoy JP, Hogarty GE, Steingard S: Optimal dose of neuroleptic in acute schizophrenia: a controlled study of neuroleptic threshold and higher haloperidol dose. Arch Gen Psychiatry 1991; 48:739–745Crossref, Medline, Google Scholar
37 Stone CK, Garver DL, Griffith J, Hirschowitz J, Bennett J: Further evidence of a dose-response threshold for haloperidol in psychosis. Am J Psychiatry 1995; 152:1210–1212Link, Google Scholar
38 Janicak P, Javaid J, Sharma R, Leach A, Easton M, Dowd S, Davis J, Dowd S, Davis J: Haloperidol dose/response relationship—results from a random-assignment, double blind study (abstract). Neuropsychopharmacology 1994; 10:106SGoogle Scholar
39 Smith RC, Chua JW, Lipetsker B, Bhattacharyya A: Efficacy of risperidone in reducing positive and negative symptoms in medication-refractory schizophrenia: an open prospective study. J Clin Psychiatry 1996; 57:460–466Crossref, Medline, Google Scholar
40 Cavallaro R, Colombo C, Smeraldi E: A pilot, open study on the treatment of refractory schizophrenia with risperidone and clozapine. Human Psychopharmacology 1995; 10:231–234Crossref, Google Scholar
41 Lacey RL, Preskorn SH, Jerkovich GS: Is risperidone a substitute for clozapine for patients who do not respond to neuroleptics? (letter). Am J Psychiatry 1995; 152:1401Medline, Google Scholar
42 Schotte A, Janssen PFM, Gommeren W, Luyten WHML, VanGompel P, Lesage AS, DeLoore K, Leysen JE: Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996; 124:57–73Crossref, Medline, Google Scholar
43 Loch C, Halldin C, Bottlaender M, Swahn CG, Moresco RM, Maziere M, Farde L, Maziere B: Preparation of [Br-76]FLB 457 and [Br-76]FLB 463 for examination of striatal and extrastriatal dopamine D-2 receptors with PET. Nucl Med Biol 1996; 23:813–819Crossref, Medline, Google Scholar

