
Increased Striatal Dopamine Transmission in Schizophrenia: Confirmation in a Second Cohort
Abstract
OBJECTIVE: The authors previously observed an increase in striatal dopamine transmission following amphetamine challenge in 15 untreated patients with schizophrenia compared to 15 matched healthy subjects. The purpose of this study was to replicate this finding in a new cohort of schizophrenic patients and healthy subjects. METHOD: Fifteen patients with schizophrenia and 15 healthy subjects matched for age, gender, ethnicity, and parental socioeconomic status were recruited for this study. Patients fulfilled DSM-IV criteria for schizophrenia, had no history of alcohol or substance abuse or dependence, and were neuroleptic free for a minimum of 21 days. Amphetamine-induced dopamine release was assessed by the reduction in dopamine D2 receptor availability induced by an acute amphetamine challenge (0.3 mg/kg, intravenous bolus). Reduction in D2 receptor availability was measured with single photon emission computed tomography and the D2 receptor radiotracer [123I]IBZM. RESULTS: No differences were observed between patients with schizophrenia and the comparison group in D2 receptor availability at baseline. Patients with schizophrenia exhibited a significantly larger reduction in D2 receptor availability following acute amphetamine challenge than the comparison group. In this study, the effect size was smaller than in the first study. Excess dopamine release following amphetamine was associated with transient emergence or worsening of positive symptoms. CONCLUSIONS: In this new cohort of subjects the authors replicated their initial observation of a dysregulation of striatal dopamine release in schizophrenia.
Conventional in vivo neuroreceptor imaging with positron emission tomography (PET) or single photon emission computed tomography (SPECT) provides noninvasive measurement of the density and affinity of a variety of neuroreceptors in the living brain. In addition, these techniques can be used to measure fluctuations in the concentration of endogenous transmitters in the vicinity of these receptors (1, 2). Competition between radiotracers and transmitters for binding to neuroreceptors is the principle underlying this application. This recent development of neuroreceptor imaging allows direct measurement of regional synaptic transmission in specific neurotransmitter systems, correlation of these measurements with behaviors and symptoms, and an exploration of the role of neurochemical imbalances in the pathogenesis of neuropsychiatric disorders.
Specifically, we developed a method to measure amphetamine-induced dopamine release with SPECT and the radiotracer [123I](S)-(-)-3-iodo-2-hydroxy-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide ([123I]IBZM), an antagonist at the D2 and D5 receptors (3). We initially observed that amphetamine challenge reduced the [123I]~IBZM binding potential in baboons. In vivo binding potential is the product of the density and affinity of available receptors, i.e., receptors not occupied by dopamine. Since amphetamine is devoid of significant affinity for D2 receptors, we postulated that this effect was mediated by increased dopamine release and displacement of [123I]~IBZM specific binding by dopamine. This mechanism was supported by the observation that pretreatment with the dopamine depleter alpha-methyl-para-tyrosine prevented the effect of amphetamine on [123I]IBZM binding potential (4). In addition, we established the existence of a good correlation between amphetamine-induced dopamine release measured with microdialysis and the amphetamine-induced decrease in [123I]IBZM binding potential measured with SPECT (4). Therefore, measuring the reduction in [123I]IBZM binding potential following amphetamine administration provides a noninvasive method to estimate the magnitude of amphetamine-induced dopamine release. Extending this method to healthy volunteers demonstrated its feasibility in humans (5).
We used this technique to study amphetamine-induced dopamine release in patients with schizophrenia. In a first cohort of 15 patients and 15 matched healthy subjects, we observed a significantly greater amphetamine-induced decrease in the [123I]IBZM binding potential in the schizophrenic group (mean=–19.5%, SD=15.7%) than in the healthy group (mean=–7.6%, SD=8.0%) (two-tailed t test, F=6.87, df=1,28, p<0.05) (6). These data provided the first in vivo evidence for a dysregulation of striatal dopamine release in schizophrenia. This result was independently replicated by Breier et al. (7) using PET, the radiotracer [11C]raclopride, and a smaller dose of amphetamine (0.2 mg/kg).
In the present investigation, we attempted to replicate these findings in a new cohort of 15 drug-free schizophrenic subjects and 15 matched healthy subjects. We kept the experimental conditions similar to those of the first study in order to allow subsequent meta-analysis of the pooled data.
METHOD
Subjects
Inclusion criteria for patients were as follows: 1) diagnosis of schizophrenia according to DSM-IV; 2) no other DSM-IV axis I diagnosis; 3) no history of alcohol or substance abuse or dependence; 4) absence of any psychotropic medication for at least 21 days before the study (with the exception of lorazepam, which was allowed at a maximal dose of 3 mg/day up to 24 hours before the study); 5) no concomitant or past severe medical conditions; 6) no pregnancy; 7) no current suicidal or homicidal ideation; and 8) ability to provide informed consent. After explanation of the nature and risks of the study, the ability of the patient to provide informed consent was formally evaluated by asking the patient to complete a multiple-choice questionnaire (available from Dr. Abi-Dargham on request). According to the recommendations of the National Alliance for the Mentally Ill (Arlington, Va.), assent from involved family members was also obtained. All patients were admitted to a research ward for the duration of the study, including the washout period.
Inclusion criteria for the comparison group were as follows: 1) absence of past or present neurological or psychiatric illnesses; 2) no concomitant or past severe medical conditions; 3) no pregnancy; and 4) ability to provide informed consent. Groups were matched for age, gender, race, and parental socioeconomic level, assessed by the Hollingshead scale (8).
Scan Protocol
SPECT experiments were carried out as previously described (5). Briefly, [123I]IBZM with specific activity >5,000 Ci/mmol and radiochemical purity >95% was prepared by direct electrophilic radioiodination of the desiodoprecursor BZM. A total mean [123I]IBZM dose of 10.7 mCi (SD=2.5) was given as a bolus (mean=4.0 mCi, SD=0.9) followed by a continuous infusion at a mean rate of 1.0 mCi/hour (SD=0.2) for the duration of the experiment (372 minutes). This protocol of administration (bolus plus constant infusion with bolus to hourly infusion rate ratio of 3.92 hours) has been shown to induce a state of sustained binding equilibrium. In the absence of amphetamine injection, both the specific and nonspecific activity remained at a constant level (within plus or minus 5%) from 150 minutes to the end of the experiment (5).
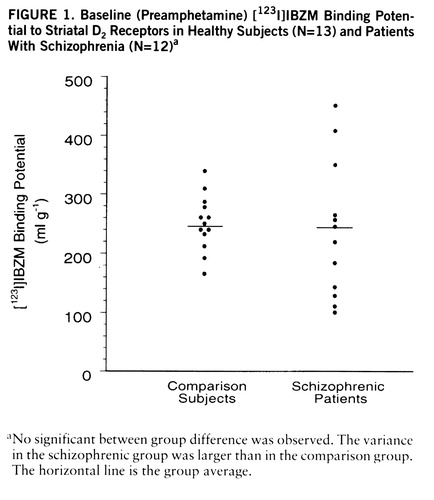
SPECT data were acquired on the PRISM 3000 (Picker, Cleveland, Ohio) with high-resolution fan beam collimators (resolution full width at half maximum, 11 mm; 123I point source sensitivity, 16.5 counts/seconds/µCi). Two scanning sessions were obtained for each subject during the course of the [123I]IBZM infusion (before and after amphetamine injection). Each scanning session consisted of nine consecutive acquisitions of 8 minutes each. The first scanning session was obtained from 180 to 252 minutes. After completion of the first scanning session, dextroamphetamine sulfate was injected intravenously at a dose of 0.3 mg/kg over 30 seconds. During the 45 minutes following the amphetamine injection, subjects were outside the scanner in order to evaluate the psychiatric response to amphetamine. The second scanning session (i.e., after amphetamine injection) was obtained from 300 to 372 minutes.
The clinical response to the amphetamine challenge was evaluated with the Positive and Negative Syndrome Scale (9). Baseline ratings were obtained 60 minutes before the first scanning session. Postamphetamine ratings were obtained 30 minutes after the injection of amphetamine (i.e., during the interval between the first and second scanning sessions). For positive and negative subscales, a change of at least 4 points from baseline was considered clinically significant.
Plasma metabolite-corrected [123I]IBZM steady-state concentration was measured by extraction followed by high pressure liquid chromatography on four venous samples collected at 20-minute intervals from 180 to 260 minutes (5). Determination of the plasma [123I]IBZM free fraction was performed by ultrafiltration (10). Plasma [123I]IBZM clearance was calculated as the ratio of steady-state concentration to infusion rate. Amphetamine plasma concentration was measured by gas chromatography on three venous samples obtained at 10, 20, and 40 minutes after amphetamine injection. To measure peripheral effects of amphetamine on dopamine metabolism, plasma levels of the dopamine metabolite homovanillic acid (HVA) were measured on six samples: three pre~amphetamine samples were collected at 20-minute intervals during the first scanning session (180, 200, and 220 minutes), and three postamphetamine samples were collected during the second scanning session (300, 320, and 340 minutes). Plasma HVA levels were assayed as previously described by gas chromatography and mass spectrometry; deuterated internal standards were used (11).
Data Analysis
SPECT images were analyzed as previously described (6). Unless otherwise specified, between-group comparisons were performed with two-tailed unpaired t tests. Relationships between continuous variables were analyzed with the Pearson product-moment correlation coefficient. A probability value of 0.05 was selected as the significance level. Effect size was calculated as the mean for schizophrenic patients minus the mean for healthy subjects divided by the standard deviation for healthy subjects. Because of lack of a second intravenous line, plasma [123I]IBZM and amphetamine were not collected for three patients and two healthy subjects.
RESULTS
Subjects
The groups did not significantly differ in age, gender, ethnicity, or parental socioeconomic status (table 1). Patients had significantly lower socioeconomic status than subjects in the comparison group (F=13.8, df=1,28, p<0.001). Two patients were experiencing a first psychotic episode and were considered neuroleptic naive (one had never received a neuroleptic; the other had less than 1 week of lifetime exposure). The other 13 patients had a mean duration of illness of 17 years (SD=7). Antipsychotics were discontinued in three patients for the purpose of participation in this study (for 22–38 days). Ten patients were noncompliant with antipsychotic treatment at the time of recruitment. For these patients, the average neuroleptic-free interval was 169 days (SD=145) (an index of 365 days was used for patients drug free for more than 1 year [N=3]). After completion of the study, all patients were offered antipsychotic medication and clinical follow-up.
Five patients received lorazepam at doses lower than 3 mg/day up to 24 hours before the scan. At the time of the scan, baseline scores were 44 (SD=11) on the Brief Psychiatric Rating Scale (BPRS) (12) and 18.5 (SD=5.1) and 19.6 (SD=7.0), respectively, on the positive and negative symptom subscales of the Positive and Negative Syndrome Scale.
Scan Parameters
As previously reported, the bolus plus constant infusion paradigm used in this study, with a bolus to hourly infusion ratio of 3.92 hours, induced steady-state [123I]IBZM plasma levels by the time of the scan. The steady-state quality of the plasma input function was evaluated by the slope of the metabolite-corrected plasma [123I]IBZM concentration from 180 to 260 minutes. This slope was small and not different between groups (schizophrenic patients: mean=–1.7%/hour, SD=7.3%; healthy subjects: mean=–2.3%/hour, SD=3.5%). Similarly, the slope of the occipital activity was negligible and did not differ between the groups (schizophrenic patients: mean=–0.5%/hour, SD=3.6%; healthy subjects: mean=–0.6%/hour, SD=2.9%). None of these slope distributions had a mean value significantly different from zero (one-sample t test), indicating that an adequate steady-state input function was achieved in both groups. Plasma [123I]IBZM clearance or free fraction did not differ between the groups (plasma [123I]IBZM clearance: schizophrenic patients, mean=63 liters/hour, SD=21; healthy subjects, mean=71 liters/hour, SD=10; plasma [123I]IBZM free fraction: schizophrenic patients, mean=3.2%, SD=0.7%; healthy subjects, mean=3.6%, SD=0.9%).
Baseline [123I]IBZM Binding Potential
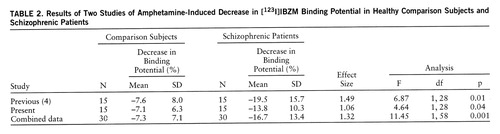
No differences were observed in baseline [123I]IBZM binding potential between patients (mean=239 ml g–1, SD=115) and healthy subjects (235 ml g–1, SD=46) (figure 1). The variability of [123I]IBZM binding potential was significantly larger in patients (F=6.10, df=11,12, p<0.005). Baseline [123I]IBZM binding potential was significantly correlated with age (r2=0.18, F=5.2, df=1,23, p<0.05), showing a mean decline of 14% (SD=6%) per decade. The interaction between age and diagnosis was not significant, showing no differences in the rate of [123I]IBZM binding potential decrease with age between schizophrenic and healthy subjects. In the schizophrenic group, baseline [123I]IBZM binding potential was not correlated with baseline clinical symptoms (total BPRS and positive and negative subscales of the Positive and Negative Syndrome Scale), the neuroleptic-free interval, or lifetime neuroleptic exposure.
Amphetamine Peripheral Effects
Amphetamine plasma levels were similar between groups (schizophrenic patients: mean=23 ng/ml, SD=7; healthy subjects: mean=23 ng/ml, SD=10). Amphetamine induced a marked but transient increase in systolic and diastolic blood pressure. No between-group differences were observed in the blood pressure response to amphetamine (peak increase in systolic blood pressure: schizophrenic patients, mean=36 mm Hg, SD=12; healthy subjects, mean=41 mm Hg, SD=14; peak increase in diastolic blood pressure: schizophrenic patients, mean=20 mm Hg, SD=24; healthy subjects, mean=18 mm Hg, SD=6). Plasma HVA levels at baseline were not different between groups (schizophrenic patients: mean=9.7 ng/ml, SD=2.7; healthy subjects: mean=9.7 ng/ml, SD=3.9). Mean postamphetamine plasma HVA levels were 3.6 ng/ml (SD=1.1) and 3.2 ng/ml (SD=0.6) in healthy subjects and schizophrenic patients, respectively. No differences between groups were observed in the reduction of plasma HVA induced by amphetamine (schizophrenic patients: mean=–60%, SD=10%; healthy subjects: mean=–63%, SD=13%).
Amphetamine Effect on [123I]IBZM Binding Potential
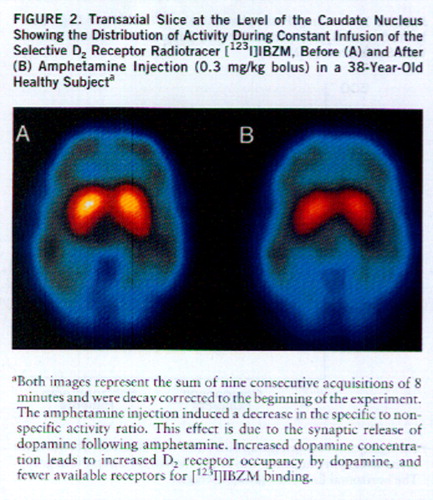
Amphetamine-induced reduction in [123I]IBZM binding potential was larger in schizophrenic patients (mean=–13.8%, SD=10.3%) than in healthy subjects (mean=–7.1%, SD=6.2%) (F=4.64, df=1,28, p<0.05) (figures 2 and 3). A nonsignificant higher variability in the response was observed in the schizophrenic group (F=2.71, df=14,14, p=0.07). No relationship between age and amphetamine-induced [123I]~IBZM displacement was observed in the two groups (patients and healthy subjects) combined (r2<0.01, N=30, p=0.71) or analyzed separately (schizophrenic patients: r2<0.01, N=15, p=0.91; healthy subjects: r2=0.02, N=15, p=0.58). No relationship was observed between amphetamine plasma levels and [123I]IBZM displacement, either in the entire study group (r2<0.01, N=30, p=0.98) or in each group considered separately (schizophrenic patients: r2<0.01, N=15, p=0.68; healthy subjects: r2=0.03, N=15, p=0.58). No correlations were observed between amphetamine-induced [123I]IBZM displacement and other indices of peripheral amphetamine activity, such as changes in systolic blood pressure (r2=0.03, N=30, p=0.33) or changes in plasma HVA (r2=0.05, N=30, p=0.28).
In the schizophrenic group, amphetamine-induced [123I]IBZM displacement was not associated with duration of the neuroleptic-free interval (r2=0.03, N=13, p=0.50), lifetime neuroleptic exposure (r2<0.01, N=13, p=0.88), duration of illness (r2=0.03, N=15, p=0.54), or the use of lorazepam in the week preceding the study (patients taking lorazepam [N=5]: mean=–20%, SD=12%; patients without lorazepam [N=10]: mean=–11%, SD=1%) (F=3.10, df=1,13, p=0.10). The two neuroleptic-naive patients had an amphetamine-induced [123I]IBZM binding potential decrease of 7% and 35%, respectively. The baseline [123I]IBZM binding potential was not associated with the amphetamine-induced [123I]IBZM reduction, either in the groups analyzed together (r2=0.04, N=30, p=0.31) or separately (schizophrenic patients: r2=0.06, N=15, p=0.40; healthy subjects: r2<0.01, N=15, p=0.85).
Relationship Between Clinical and Biochemical Response
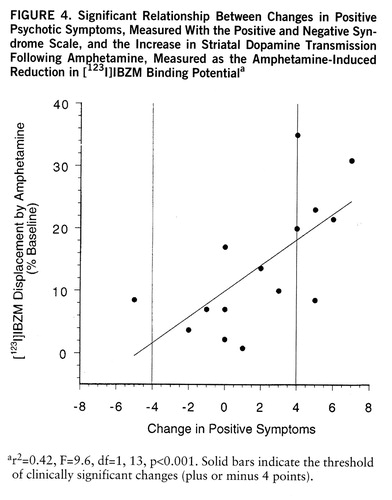
No psychotic reaction was observed in the comparison group (healthy subjects). In the schizophrenic group, six patients experienced a psychotic reaction to the amphetamine injection (i.e., increase of 4 or more points on the positive subscale of the Positive and Negative Syndrome Scale), eight patients showed no significant change in positive symptoms, and one patient significantly improved (i.e., decrease of 4 or more points on the positive subscale of the Positive and Negative Syndrome Scale). Amphetamine-induced dopamine release was significantly larger in the patients who had a psychotic reaction (mean=–23%, SD=9%, N=6) than in the patients who did not have a psychotic reaction (mean=–8%, SD=5%, N=9) (F=16.6, df=1,13, p<0.005). The increase in positive symptoms was significantly correlated with the reduction in [123I]IBZM binding potential (r2=0.42, F=9.6, df=1,13, p<0.001) (figure 4). Baseline [123I]IBZM binding potential was not different between schizophrenic patients who had a psychotic reaction (mean=216 ml g–1, SD=95, N=6) and those who did not (mean=255 ml g–1, SD=132, N=9). The severity of positive symptoms at baseline was not associated with the magnitude of the amphetamine effect on positive symptoms (r2=0.03, N=15, p=0.91) or [123I]IBZM displacement (r2=0.07, N=15, p=0.32).
Eleven patients showed no significant changes in negative symptoms following the amphetamine challenge, and four patients showed a clinically significant improvement (i.e., decrease of 4 or more points on the negative subscale of the Positive and Negative Syndrome Scale). No significant relationship was observed between improvement in negative symptoms and [123I]IBZM displacement (r2=0.07, N=15, p=0.31). Amphetamine-induced changes in positive and negative symptoms were not correlated (r2<0.01, N=15, p=0.76).
Comparison With First Study
Results in this second cohort of subjects are comparable to the results obtained in the first cohort (6) (table 2). Both cohorts were relatively similar in terms of clinical and demographic composition. In each study, no between-group differences were observed in baseline [123I]IBZM binding potential. In comparison subjects, the amphetamine effects on [123I]IBZM binding potential were identical in studies 1 and 2. In both studies, the amphetamine effect on [123I]IBZM binding potential was larger in schizophrenic patients than in healthy subjects. However, in this study, the effect size was lower (1.06) than in the first study (1.49), and the significance level of the group difference was lower (p=0.04 for this study and p=0.01 for the first study). Since the study protocols were identical, results of both studies can be combined. When the results were combined, the effect size was 1.32 and the p value was 0.001 (F=11.4, df=1,58, p=0.001).
DISCUSSION
In a new cohort of 15 schizophrenic subjects and 15 matched healthy subjects, we replicated the observation of a larger decrease in [123I]IBZM binding potential following amphetamine challenge in schizophrenic patients than in healthy subjects. Findings from these two studies, together with the independent replication by Breier et al. (7) and the preliminary results recently presented by a third group (13), clearly indicate abnormal reactivity of dopaminergic transmission to amphetamine exposure in the brain of schizophrenic subjects. We also replicated the observation that this excessive reaction is associated with behavioral consequences, i.e., worsening or emergence of psychotic symptoms.
As in the first study, the increased stimulation of dopamine transmission observed in the striata of schizophrenic patients could not be attributed to differences in plasma amphetamine levels. The measurement of plasma HVA before and after amphetamine was a new feature of this study. Since HVA is formed mainly inside the cell, the decrease in plasma HVA following amphetamine is generally attributed to depletion of the dopamine cytoplasmic pool (14–16). Thus, the decrease in plasma HVA is an indirect measure of the intracellular effect of amphetamine in the peripheral nervous system. The similarity in plasma HVA response to amphetamine indicates that the intracellular bioavailability of amphetamine was comparable between the groups. Thus, the observed group differences in striatal dopamine response to amphetamine exposure are related to central nervous system factors.
As in the first study, we could not attribute the increased dopamine response in schizophrenic patients to prior neuroleptic exposure, because of the lack of association between the amphetamine effect and previous neuroleptic exposure or duration of neuroleptic interval. These results indicate that the effect is unlikely to represent a long-term effect of previous neuroleptic medication. Similarly, we were unable to link the effect with lorazepam exposure during the week preceding the study. In conclusion, the effect seems to be associated with the illness process per se.
Abnormal dopaminergic transmission has been suspected in schizophrenia for a long time. These studies represent the first direct demonstration of abnormal regulation of striatal dopaminergic transmission in schizophrenia. The increase in dopaminergic transmission following amphetamine might be due to an increased affinity of D2 receptors for dopamine (for example, due to a higher proportion of high versus low affinity sites), a larger or more prolonged dopamine release (i.e., presynaptic factors), or a combination of both. Additional studies are needed to better characterize the exact mechanisms responsible for this increased transmission (4, 6).
Several limitations of this study should be discussed. This method has a relatively low sensitivity. We previously examined in primates the relationship between increase in extrastriatal dopamine, measured with microdialysis, and decrease in [123I]IBZM binding potential, measured with SPECT. An increase of 45% above baseline of striatal dopamine is needed to decrease [123I]IBZM binding potential by 1% (4). A similar ratio (44:1) has been reported by Breier et al. (7) for [11C]~raclopride. In a previous study, we showed that in the absence of amphetamine, the reproducibility of [123I]~IBZM binding potential measurement had an average variability (test minus retest divided by the average) of plus or minus 3.3% (SD=1.7%, range=5% to –5%). The low signal to noise ratio of the method explains why two comparison subjects had an apparent increase in [123I]IBZM binding potential following amphetamine. On the other hand, the low sensitivity of the method implies that a very large difference in synaptic dopamine increase between patients and comparison subjects corresponded to the measured differences in [123I]IBZM binding potential decrease.
Another important limitation of the method is that it does not provide any information about the baseline dopamine concentration, i.e., concentration in the absence of amphetamine. In this study we measured only the relative increase in dopamine concentration resulting from the amphetamine challenge. The observation of a normal D2 receptor availability at baseline does not imply that baseline dopamine levels are unaltered in schizophrenia (17). A study based on rapid induction of dopamine depletion with alpha-methyl-para-tyrosine is currently being conducted in our group to address this issue (18).
Finally, the low number of patients not previously exposed to antipsychotics makes it difficult to rule out the possibility that the observed effect might represent a long-term consequence of prolonged neuroleptic exposure. More neuroleptic-naive patients should be studied to address this important question.
The effect size in the second cohort (1.06) was lower than that in the first cohort (1.49). The reasons for the lower effect size are unclear. The scan protocol was identical, comparison subjects had similar effects (table 2), and the clinical characteristics of the patients were similar (tables 1 in this article and in reference 6). Therefore, it is likely that the difference in effect size is due to a sampling effect, possibly related to the large variance of the amphetamine effect in the schizophrenic group.
The mechanism, specificity, significance, and etiology of this increased dopaminergic response in schizophrenia remains to be elucidated. However, recent preclinical studies do offer interesting hypotheses. Neonatal hippocampal lesions in rodents and primates provide a potential neurodevelopmental animal model for some clinical and biochemical features of schizophrenia (19). Regulations of striatal dopamine release were compared with microdialysis between adult rhesus monkeys who underwent ablation of the amygdala-hippocampal formation within 3 weeks of birth, comparison monkeys, and monkeys who underwent the same lesion ablation during adulthood (20). Local perfusion of amphetamine in the prefrontal cortex induced a decrease in striatal dopamine release in comparison monkeys and in monkeys with adult lesions. This observation is consistent with the inhibition of subcortical dopamine by prefrontal dopamine (21). In contrast, in monkeys with neonatal lesions, prefrontal amphetamine perfusion induced an increase in striatal dopamine release, i.e., a reversal of the normal effect (inhibition). Thus, a neonatal hippocampal lesion does affect prefrontal inhibition of striatal dopamine release. The relevance of this model to schizophrenia remains to be established. Brain imaging techniques now provide the tools to directly test these preclinical models in patients.
In conclusion, we replicated in this second cohort our previous observation that schizophrenia is associated with an increased dopamine transmission following acute amphetamine challenge (6). The results of both studies were very similar, with the exception that the effect size in the second study was less pronounced than that in the first study. These studies, as well as similar results provided by other groups (7, 13), are consistent in documenting this excessive neurochemical response and provide direct evidence to support the hypothesis of a dysregulation of central dopamine transmission in schizophrenia.
 |
 |
Presented in part at the annual meeting of the Society for Neuroscience, Washington D.C., Nov. 16–21, 1996, and at the American College of Neuropsychopharmacology, Puerto Rico, Dec. 9–13, 1996. Received July 30, 1997; revision received Dec. 4, 1997; accepted Dec. 16, 1997. From the Departments of Psychiatry and Diagnostic Radiology, Yale University, New Haven; and the VA Medical Center, West Haven, Conn. Address reprint requests to Dr. Abi-Dargham, New York State Psychiatric Institute, Unit 28, 177 West 168th St., New York, NY, 10032; [email protected] (e-mail). Supported by National Alliance for Research in Schizophrenia and Depression Young Investigator Award (Dr. Laruelle), NIMH grants MH-54192 (Dr. Laruelle) and MH-30929 (Dr. Bowers), and the Veterans Administration. The authors thank Christine Fingado, Donna M. Damon, Melyssa K. Madrak, Louis A. Amici, Richard Weiss, Lynn Pantages-Torok, and the staff of the NeuroSPECT Laboratory at Yale-New Haven Hospital for technical assistance and the staff of the Neuropsychiatric Studies Unit at the West Haven VA Medical Center for the clinical care of the patients.
1 Logan J, Dewey SL, Wolf AP, Fowler JS, Brodie JD, Angrist B, Volkow ND, Gatley SJ: Effects of endogenous dopamine on measures of [18F]N-methylspiroperidol binding in the basal ganglia: comparison of simulations and experimental results from PET studies in baboons. Synapse 1991; 9:195–207Crossref, Medline, Google Scholar
2 Innis RB, Malison RT, Al-Tikriti M, Hoffer PB, Sybirska EH, Seibyl JP, Zoghbi SS, Baldwin RM, Laruelle MA, Smith E, Char~ney DS, Heninger G, Elsworth JD, Roth RH: Amphetamine-stimulated dopamine release competes in vivo for [123I]IBZM binding to the D2 receptor in non-human primates. Synapse 1992; 10:177–184Crossref, Medline, Google Scholar
3 Kung HF, Kasliwal R, Pan S, Kung M-P, Mach RH, Guo Y-Z: Dopamine D-2 receptor imaging radiopharmaceuticals: synthesis, radiolabeling, and in vitro binding of R-(+)- and S-(-)-3-iodo-2-hydroxy-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide. J Med Chem 1988; 31:1039–1043Crossref, Medline, Google Scholar
4 Laruelle M, Iyer RN, Al-Tikriti MS, Zea-Ponce Y, Malison R, Zoghbi SS, Baldwin RM, Kung HF, Charney DS, Hoffer PB, Innis RB, Bradberry CW: Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse 1997; 25:1–14Crossref, Medline, Google Scholar
5 Laruelle M, Abi-Dargham A, van Dyck CH, Rosenblatt W, Zea-Ponce Y, Zoghbi SS, Baldwin RM, Charney DS, Hoffer PB, Kung HF, Innis RB: SPECT imaging of striatal dopamine release after amphetamine challenge. J Nucl Med 1995; 36:1182–1190Medline, Google Scholar
6 Laruelle M, Abi-Dargham A, Van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB: Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA 1996; 93:9235–9240Crossref, Medline, Google Scholar
7 Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, deBartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D: Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 1997; 94:2569–2574Crossref, Medline, Google Scholar
8 Hollingshead AB: Four-Factor Index of Social Status. New Haven, Conn, Yale University, Department of Sociology, 1975Google Scholar
9 Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276Crossref, Medline, Google Scholar
10 Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, Innis RB: Evaluation of ultrafiltration for the free fraction determination of single photon emission computerized tomography (SPECT) radiotracers: β-CIT, IBF and iomazenil. J Pharm Sci 1994; 83:1014–1019Crossref, Medline, Google Scholar
11 Bacopoulos NG, Heninger G, Roth RH: Effects of haloperidol and probenecid on plasma and CSF dopamine metabolites in the rhesus monkey (Macacca mulatta). Life Sci 1978; 23:1805–1812Crossref, Medline, Google Scholar
12 Overall JE, Gorham DR: The Brief Psychiatric Rating Scale. Psychol Rep 1962; 10:799–812Crossref, Google Scholar
13 Wong D, Gjedde A, Reith J, Grunder G, Szymanski S, Yokoi F, Hong C, Nestadt G, Neufeld K, Pearlson G, Tune L, Angrist B: Multiple lines of evidence for dopamine dysfunction in psychosis by imaging (abstract). Schizophr Res 1997; 24:183Crossref, Google Scholar
14 Zetterstrom T, Sharp T, Marsden CA, Ungerstedt U: In vivo measurement of dopamine and its metabolites by intracerebral dialysis: changes after d-amphetamine. J Neurochem 1983; 41:1769–1773Crossref, Medline, Google Scholar
15 Zetterstrom T, Sharp T, Ungerstedt U: Further evaluation of the mechanism by which amphetamine reduces striatal dopamine metabolism: a brain dialysis study. Eur J Pharmacol 1986; 132:1–9Crossref, Medline, Google Scholar
16 Kuczenski R, Segal D: Concomitant characterization of behavioral and striatal neurotransmitter response to amphetamine using in vivo microdialysis. J Neurosci 1989; 9:2051–2065Crossref, Medline, Google Scholar
17 Farde L, Wiesel F, Stone-Elander S, Halldin C, Nordström AL, Hall H, Sedvall G: D2 dopamine receptors in neuroleptic-naive schizophrenic patients: a positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry 1990; 47:213–219Crossref, Medline, Google Scholar
18 Hietala J, SyvÄlahti E, Vuorio K, Någren K, Lehikoinen P, Ruotsalainen U, RÄkkölÄinen V, Lehtinen V, Wegelius U: Striatal D2 dopamine receptor characteristics in neuroleptic-naive schizophrenic patients studied with positron emission tomography. Arch Gen Psychiatry 1994; 51:116–123Crossref, Medline, Google Scholar
19 Lipska B, Weinberger D: Cortical regulation of the mesolimbic dopamine system: implication for schizophrenia, in Limbic Motor Circuits and Neuropsychiatry. Edited by Kalivas PW, Barnes CD. Boca Raton, Fla, CRC Press, 1993, pp 329–349Google Scholar
20 Kolachana BS, Saunders RC, Bachevalier J, Weinberger DR: Abnormal prefrontal cortical regulation of striatal dopamine release after neonatal medial temporal-limbic lesions in rhesus monkeys. Abstracts of the Society for Neuroscience 1996; 22:1974Google Scholar
21 Deutch AY: Prefrontal cortical dopamine systems and the elaboration of functional corticostriatal circuits: implications for schizophrenia and Parkinson's disease. J Neural Transm 1993; 91:197–221Crossref, Google Scholar

