
Senile Degeneration and Cognitive Impairment in Chronic Schizophrenia
Abstract
Objective:This study was an investigation of the role of Alzheimer-type senile degenerative abnormalities in the cognitive impairment of chronic schizophrenia.Method:The study group comprised 145 deceased elderly institutionalized psychiatric patients: 66 with schizophrenia, 26 with mood disorders, 36 with dementia, and 17 with other psychiatric diagnoses. The comparison group included 16 deceased elderly individuals without neurologic or psychiatric disease. Psychiatric diagnoses and cognitive status were established by standardized review of medical records. Neuritic senile plaques and neurofibrillary tangles were identified immunohistochemically and counted, by investigators blind to clinical information, in standardized regions of each brain.Results:Of the subjects with schizophrenia, 68% had definite cognitive impairment, but only 8% satisfied neuropathological criteria for Alzheimer’s disease. Among the schizophrenia subjects without Alzheimer’s disease, definite cognitive impairment was associated with higher levels of plaques and tangles. The schizophrenia subjects without definite cognitive impairment had fewer plaques and tangles than the unimpaired nonpsychiatric subjects. Conclusions:Most cases of cognitive impairment in schizophrenia could not be attributed to Alzheimer’s disease. An association of mild Alzheimer-type pathology with definite cognitive impairment was unique to schizophrenia. Enhanced sensitivity to the effects of aging on the brain may be a manifestation of diminished cognitive reserve in schizophrenia. Am J Psychiatry 1998; 155: 1536-1543
Cognitive impairment is a well-recognized element of schizophrenia, not simply attributable to psychotic symptoms (1, 2). Subtle impairment can sometimes be detected before the onset of frank psychosis, it is a prominent feature of schizophrenia after the onset of psychosis, and it increases in severity and prevalence with aging (3–5).
Much of the cognitive impairment associated with age in the nonpsychotic population is attributable to Alzheimer’s disease. A number of studies have tested for a high prevalence of neuropathologically verified Alzheimer’s disease in schizophrenia, with inconclusive but mostly negative results (6–11). However, just as individuals with greater educational and occupational attainments may possess a “cognitive reserve” that mitigates the cognitive effects of senile degeneration (12–15), the converse may also hold. In schizophrenia, where intellectual functions may already be impaired, low levels of neuritic senile plaques and neurofibrillary tangles, consistent with “normal aging” in a healthy individual, may be sufficient to cause recognizable cognitive decline.
In the present study we sought to evaluate this possibility by addressing three questions: 1) Is the frequency of neuropathologically defined Alzheimer’s disease higher than normal in schizophrenia? 2) Are levels of neuritic senile plaques and neurofibrillary tangles below the threshold for Alzheimer’s disease related to cognitive impairment in schizophrenia? 3) If so, does the relationship differ from that in other populations?
METHOD
Subjects
Psychiatric cases were collected from autopsies performed at seven New York State psychiatric hospitals between 1982 and 1993. An age below 45 was the only exclusion criterion. Patients were divided into four DSM-III-R diagnostic categories, on the basis of a standardized review of medical records (to be described): “schizophrenia” included all patients with a primary diagnosis of schizophrenia or schizoaffective disorder, “mood disorder” included all individuals with a primary diagnosis of major depression or bipolar disorder, “dementia” comprised all individuals with a primary diagnosis of dementia, and “other psychiatric” included all individuals with psychiatric diagnoses that did not fit into the preceding categories.
We also included a nonpsychiatric group culled from autopsies at a general hospital. Clinical records were reviewed in the same manner as for the psychiatric cases. Only those with no psychiatric diagnosis or with adjustment disorder as the only psychiatric diagnosis were included. Summaries of age and sex are given in table 1. In all cases, consent for autopsy was obtained.
The brains were preserved in 10% phosphate-buffered formalin before and after routine neuropathological examination. The data in this study were based on a second, standardized neuropathological examination (see the following), typically performed several years after the autopsy, on new paraffin blocks prepared in a vacuum-infiltration tissue processor (Instrumentation Laboratory, Lexington, Mass.). The intervals between death and autopsy and between autopsy and the standardized neuropathological examination are included in table 1.
Clinical Evaluations
Hospital records were reviewed independently by at least two members of a team of psychiatrists and psychologists, who were blind to autopsy results. Consensus diagnoses were determined after discussion of these reviews by this entire team. All raters were trained in the chart review protocol and the modified Diagnostic Evaluation After Death and were tested for interrater reliability (16). The diagnostic procedures followed DSM-III-R guidelines, except that cognitive loss in the context of schizophrenia was not diagnosed as a separate dementia.
Review with the modified Diagnostic Evaluation After Death also evaluated cognitive impairment. Cases were rated as showing “definite cognitive impairment” on the basis of straightforward evidence of a decline in cognitive function, without recovery, in the form of 1) psychometric test results, 2) clinical mental status examinations, or 3) a general level of functioning consistent with any dementia diagnosis. In most cases, ratings of definite cognitive impairment were based on physicians’ notes of significantly impaired memory or orientation and on staff notes of declines in several areas of self-care. While meeting the DSM criteria for dementia was not a formal requirement, most subjects classified as having definite cognitive impairment would meet them. We required a period of at least 1 year of impaired functioning without recovery. Cognitive impairment was not diagnosed in the context of general physical decline within the last year of life. A final rating of definite cognitive impairment indicated a consensus that these criteria were definitely satisfied.
Evaluation of Neuritic Senile Plaques and Neurofibrillary Tangles
Neuritic senile plaques and neurofibrillary tangles were evaluated in the following regions: superior and middle frontal gyri, at the level of the crossing of the anterior commissure; hippocampal formation and adjacent neocortex at the levels of the amygdala, pes hippocampi, and lateral geniculate body; superior and middle temporal gyri at the level of the lateral geniculate body; inferior parietal lobule at the level of the posterior end of the pulvinar; and calcarine and adjacent cortex approximately 1 cm rostral to the caudal tip of the occipital lobe. The single field of highest density was counted on each slide (17, 18) except the hippocampal slides, where the allocortex and neocortex were treated separately. Only neuritic plaques were counted (18) since amyloid plaques without neuritic elements may be very numerous in the absence of dementia (19, 20). Counts were made with a 25× objective, which afforded a field of 0.38 mm2. (We made appropriate adjustments when applying the Khachaturian criteria [17], defined for 1 mm2.) Neuritic senile plaques were identified by immunohistochemistry with Alz 50, which stains the neuritic elements in the plaques but not β-amyloid (21). Neurofibrillary tangles were identified by immunohistochemistry with Ab 39, which recognizes paired helical filaments (22). Details of the staining techniques and confirmation of their applicability to archival, formalin-fixed material are described elsewhere (23). The right and left hemispheres were randomly alternated for sampling.
All microscopic examinations were performed by investigators who were blind to any clinical information and to the source of the specimen. Neuritic senile plaques and neurofibrillary tangles were counted manually by a single observer (D.L.). For evaluations other than the fulfillment of the Khachaturian criteria for Alzheimer’s disease (17), counts of neuritic senile plaques and neurofibrillary tangles were averaged separately across the allocortical and neocortical regions for each case.
Analysis of Data
First, we determined the prevalence of Alzheimer’s disease in each diagnostic group by applying the Khachaturian age-dependent neuropathological criteria (17).
Second, we compared the numbers of neuritic senile plaques and neurofibrillary tangles in the schizophrenia subjects with and without definite cognitive impairment, by two-tailed t tests.
Third, we used analysis of covariance (ANCOVA) to compare the age-adjusted levels of neuritic senile plaques and neurofibrillary tangles in the schizophrenia subjects without definite cognitive impairment and the nonpsychiatric subjects (all of whom lacked definite cognitive impairment, by definition). This tested for lower tolerance to neuritic senile plaques or neurofibrillary tangles in schizophrenia.
Fourth, we compared the schizophrenia and mood disorder subjects, using chi-square analysis to compare rates of cognitive impairment. We used logistic regression to test the effect of diagnosis on cognitive impairment while controlling for age, neuritic senile plaques, and neurofibrillary tangles.
RESULTS
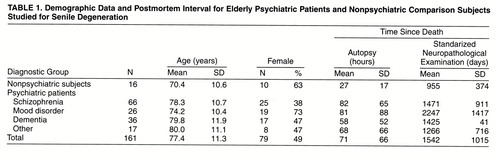
Definite cognitive impairment was present in 68% of the schizophrenia subjects (N=45), 19% of the mood disorder subjects (N=5), and 59% of the “other psychiatric” subjects (N=10). By definition, definite cognitive impairment was absent in the nonpsychiatric subjects and was present in all of the dementia subjects. As expected, neuritic senile plaques and neurofibrillary tangles were significantly more numerous in the dementia group than in any of the other diagnostic groups (figure 1).
The various measures of senile degeneration were highly correlated with one another (table 2).
Frequency of Neuropathological Alzheimer’s Disease in Schizophrenia
Five (8%) of the schizophrenia subjects (aged 73, 80, 80, 82, and 89), all with definite cognitive impairment, and 17 (47%) of the dementia subjects met the Khachaturian neuropathological criteria for Alzheimer’s disease (17). The counts of neuritic senile plaques and of neurofibrillary tangles were similar for the subjects with Alzheimer’s disease in the two diagnostic groups. Alzheimer’s disease can thus explain cognitive impairment in only 11% of the 45 schizophrenia subjects with definite cognitive impairment.
Comparison of Schizophrenia Subjects With and Without Definite Cognitive Impairment
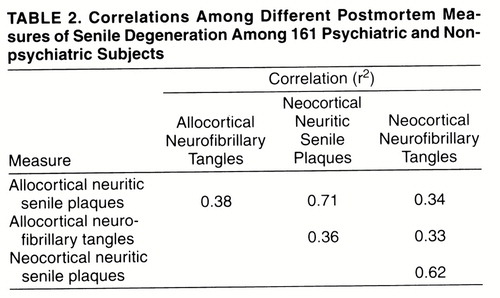
The schizophrenia subjects with and without definite cognitive impairment were similar in terms of duration of illness (without impairment: mean=46.2 years, SD=12.0, range=27–72; with impairment: mean=54.8, SD=8.9, range=25–72) and history of somatic treatments (table 3). The schizophrenia subjects with cognitive impairment were significantly older (mean age=80.6 years, SD=10.3) than those without cognitive impairment (mean=73.3, SD=9.8) (t=2.72, df=64, p=0.008); all but one of the 17 schizophrenia subjects aged 85 or older were cognitively impaired. With the subjects aged 85 or older eliminated, the mean ages of the schizophrenia groups with cognitive impairment (mean=75.2, SD=8.3) and without cognitive impairment (mean=72.6, SD=9.4) were similar (t=1.01, df=47, p=0.32), while all measures of neuritic senile plaques and neurofibrillary tangles were greater in the group with definite cognitive impairment. The differences were statistically significant for all measures except neocortical neurofibrillary tangles. When the schizophrenia subjects with neuropathological Alzheimer’s disease were excluded, the difference in neuritic senile plaques remained significant (figure 2). With schizophrenia subjects of all ages included, the differences were larger.
The most specific neuropathological predictor of definite cognitive impairment in schizophrenia was neocortical neuritic senile plaques. All but three of the schizophrenia subjects with any neocortical neuritic senile plaques had definite cognitive impairment, and each of these three had only one neuritic senile plaque per field in only one section. However, neocortical neuritic senile plaques were not a sensitive predictor: 13 of the 30 schizophrenia subjects below age 85 with no neocortical neuritic senile plaques had definite cognitive impairment. Among these 30 cases without neocortical neuritic senile plaques, those with and without definite cognitive impairment did not differ significantly on any of the other measures of neuritic senile plaques or neurofibrillary tangles.
There were 14 schizophrenia subjects, all with definite cognitive impairment, who did not have neuropathological Alzheimer’s disease but who had more than one neocortical neuritic senile plaque per field in at least one section or had one neocortical neuritic senile plaque per field in more than one section. One had frontal and parietal neuritic senile plaques, seven had only temporal neocortical neuritic senile plaques, and six had temporal neocortical neuritic senile plaques plus neuritic senile plaques in the frontal (N=3), parietal (N=3), and/or occipital (N=1) neocortex.
Comparison of Schizophrenia Subjects Without Definite Cognitive Impairment and Nonpsychiatric Subjects
Mean age-adjusted counts of neuritic senile plaques and neurofibrillary tangles were lower for the schizophrenia subjects without definite cognitive impairment than for the nonpsychiatric group, by factors ranging from about one-half (0.551 for allocortical neurofibrillary tangles) down to about one-thirtieth (0.035 for neocortical neuritic senile plaques). All differences were statistically significant except for allocortical neurofibrillary tangles (figure 3).
Comparison of Schizophrenia and Mood Disorder Subjects
Schizophrenia and mood disorder subjects below age 85 had similar mean ages (mean=74.1, SD=8.7, and mean=70.5, SD=7.5, respectively) (t=1.66, df=68, p=0.10) and similar levels of allocortical and neocortical neuritic senile plaques and neurofibrillary tangles (not shown; similar to levels for all ages, figure 1). Both groups had chronic illnesses (schizophrenia: mean duration=48.9 years, SD=9.6, range=25–63; mood disorder: mean=27.7 years, SD=6.0, range=5–55), and they received various somatic treatments with similar frequencies (table 3). However, definite cognitive impairment was over six times as prevalent in the schizophrenia group (29 of 49) as in the mood disorder group (two of 21) (χ2=14.7, df=1, p=0.0001). When we adjusted for age, neurofibrillary tangles, and neuritic senile plaques, the odds ratio for definite cognitive impairment in schizophrenia versus mood disorders was 12.7 (95% confidence interval=2.1–76.7). Thus, cognitive function appears to be more vulnerable to the effects of neuritic senile plaques and neurofibrillary tangles in schizophrenia than in mood disorders.
Among the schizophrenia subjects under age 85, those with cognitive impairment had fewer years of schooling (7.8 years versus 10.2 years for those without cognitive impairment) (t=2.47, df=47, p=0.02). The mood disorder subjects under age 85 had a slightly higher mean educational level than the schizophrenia subjects under age 85 (10.2 versus 8.8 years) (t=1.67, df=67, p=0.10). When education was included in the logistic regression, the odds ratio for cognitive impairment in schizophrenia versus mood disorders was reduced to 10.7 (95% confidence interval=1.6–70.2).
Other Neuropathological Explanations for Dementia
Only six schizophrenia subjects (aged 76–81) had other neuropathological conditions that sometimes cause cognitive impairment: four had Parkinson’s disease, one had hydrocephalus of unknown etiology, and one had a small (<1 cm diameter) focus of idiopathic fibrosis in the left frontal lobe. Exclusion of these cases produced little change in the number of neuritic senile plaques or neurofibrillary tangles in the schizophrenia subjects with or without cognitive impairment.
Among the 36 dementia subjects, there were 17 cases of Alzheimer’s disease, seven cases of cerebral infarct, two each of frontal lobe dementia, infection, and metabolic disorders, one case of Huntington’s disease, and one case of neuroaxonal dystrophy. The remaining four cases (11%) showed no significant pathology except for Alzheimer-type changes of insufficient severity to meet the age-dependent criteria for a histological diagnosis of Alzheimer’s disease (17).
DISCUSSION
Our study yielded four main findings.
| 1. | While definite cognitive impairment was present in 68% of a group of elderly, chronically institutionalized individuals with schizophrenia, neuropathological Alzheimer’s disease was present in only 8%. | ||||
| 2. | Schizophrenia subjects with definite cognitive impairment had more neuritic senile plaques (even after exclusion of cases with neuropathological Alzheimer’s disease) than did schizophrenia subjects without cognitive impairment. | ||||
| 3. | Schizophrenia subjects without definite cognitive impairment had fewer neuritic senile plaques and neurofibrillary tangles than did cognitively intact nonpsychiatric patients. | ||||
| 4. | Cognitive impairment was much more common among schizophrenia subjects than among chronically institutionalized mood disorder patients, while the two groups were comparable in terms of age, Alzheimer-type changes, and somatic treatments. | ||||
A parsimonious interpretation of these findings is that schizophrenia creates a vulnerable state in which even mild senile degeneration, particularly in the form of neocortical neuritic senile plaques, is sufficient but not necessary to cause definite cognitive impairment. In contrast, mild levels of senile degenerative change appear to be better tolerated in subjects without preexisting psychiatric disease. For example, four of 16 normal subjects had at least four neuritic senile plaques per field in at least one neocortical region. However, while the nonpsychiatric subjects clearly lacked the behavioral manifestations of cognitive impairment that were present in the subjects with schizophrenia, neuropsychological testing or detailed clinical evaluation may elicit subtle cognitive differences between elderly subjects with and without mild Alzheimer-type changes (20, 24, 25). Individuals who can tolerate such cognitive loss without behavioral manifestations must posses either compensatory abilities or a surfeit of cognitive ability. We suggest that these are lacking in chronically institutionalized subjects with schizophrenia.
Our findings are similar to those of El-Mallakh et al. (26), who found more frontal and hippocampal senile plaques in 10 intellectually impaired schizophrenia subjects (mean age=84 years) than in seven intellectually intact schizophrenia subjects (mean age=62). In striking similarity to our study, the mean plaque counts for the intellectually intact schizophrenia subjects were extremely low, approximately one-tenth of the mean value for seven normal subjects (mean age=73).
Two recent studies of elderly, chronic inpatients had findings somewhat different from ours. One study (10) showed levels similar to ours for neocortical neuritic senile plaques in nonpsychiatric subjects and cognitively impaired schizophrenia subjects but a higher value for cognitively unimpaired schizophrenia subjects, close to the value for the nonpsychiatric subjects. All ages were similar, and we cannot attribute this difference to staining or counting procedures, since the values were similar for the first two groups, nor to overdiagnosis of definite cognitive impairment on our part, since Purohit et al. (10) classified 87 of 100 schizophrenia subjects as cognitively impaired, while we classified 68% as having definite cognitive impairment. Purohit et al. counted neuritic senile plaques in the orbitofrontal cortex, while we did not. If their higher value was attributable to neuritic senile plaques in this region, it is possible that these are better tolerated than neuritic senile plaques in the temporal neocortex. In another study (27) there was no correlation between any neuropathological measure and any cognitive measure in 23 elderly schizophrenia subjects. Since that study did not include counts of neuritic plaques, it is difficult to compare with ours.
Enhanced sensitivity of cognition to neuritic senile plaques was not apparent in our group of chronically institutionalized patients with mood disorder, although this group was similar to the schizophrenia group in terms of age, presence of psychosis, institutionalization, and somatic treatments. The subjects in the two groups died during the same period and in the same institutions; hence, one should expect similar reporting of their symptoms in the medical records. Thus, from the available data, we cannot attribute the high rate of cognitive impairment in schizophrenia to institutionalization, somatic treatments, or reporting biases of clinicians.
Limitations
There are several potential limitations to this study.
| 1. | The tissues had been in formalin for several years. However, we verified previously that immunoreactivity with the antibodies we used is preserved (23), and at least one other study of immunohistochemistry for senile degenerative changes has used such tissue (28). Furthermore, our rates of neuropathological Alzheimer’s disease among inpatients with schizophrenia or primary dementia are comparable to those in other studies (9, 10). | ||||
| 2. | In order to apply standardized, age-related neuropathological criteria for Alzheimer’s disease (17), we counted neuritic senile plaques and neurofibrillary tangles only in the field of highest density within each region. Thus, the counts are not necessarily representative of average densities over broad regions. However, we were very consistent in the selection of regions for examination, and the counts represent the maximum severity of pathology in each region. Hence, comparability across cases was maintained. This strategy allowed us to make the critical distinction between cases with low levels of neocortical neuritic senile plaques and those with none. | ||||
| 3. | Since “definite cognitive impairment” represents rater certainty of cognitive impairment, inadequate documentation could theoretically result in a false negative rating. This seems unlikely, however, since the records were extensive and detailed, and our rate of definite cognitive impairment (73% in schizophrenia patients aged 65 and over) is comparable to the rate of dementia (83%) in a series of live schizophrenia patients from one of the contributing institutions (29). False negative classifications would anyway work against our findings by inflating counts of neuritic senile plaques and neurofibrillary tangles in the group without definite cognitive impairment. | ||||
| 4. | All of the schizophrenia subjects died in state institutions after 1981. Therefore, they may have had unusually severe forms of the illness. | ||||
“Cognitive Reserve” in Schizophrenia
Our findings suggest that schizophrenia and senile degeneration are synergistic in producing cognitive decline. This interpretation is consistent with theories of “cognitive reserve” protecting against dementia, first proposed by Katzman et al. (30). These investigators found that the brains of nondemented elderly individuals with large numbers of neuritic senile plaques but no dementia were heavier and contained more neurons than those of elderly individuals with dementia. Evidence that higher educational and occupational levels protect against the dementing effects of neuritic senile plaques and neurofibrillary tangles has been interpreted as further support for the cognitive reserve hypothesis (14, 15). The observation that neocortical synaptic density provided the best neuroanatomic correlate for cognitive function in Alzheimer’s disease (31, 32) has led to the hypothesis that the protective effect of education resides in increased synaptic density (14). Under this model, we postulate that senile impairment of cognition in schizophrenia and nonschizophrenia subjects alike results from a continuous, age-related process that is correlated with the appearance of neuritic senile plaques. The threshold at which this process produces clinical evidence of cognitive change is hypothetically lower in individuals with schizophrenia than in others.
The underlying vulnerability in schizophrenia may be developmental; several investigators have reported low cognitive capacity in young subjects (3, 33, 34). Vulnerability could also be associated with some progressive process, yet unidentified, that is specifically related to schizophrenia. Theoretically, such a process could produce cognitive impairment by itself when sufficiently advanced, or at an earlier stage if neocortical neuritic senile plaques are also present.
Processes contributing to diminished cognitive reserve in schizophrenia have yet to be identified. In contrast to Alzheimer’s disease, where loss of synaptic density (31, 32) and cholinergic markers (35) are prominent features and correlated with cognitive impairment, diminished synaptic density has not been reported in schizophrenia, and cholinergic markers are normal (26, 35). However, reported abnormalities in cortical synaptic proteins (36–42), microtubule-associated proteins (43–45), neuronal size (46–48), and neuropil volume (49) suggest the possibility of impaired synaptic function or remodeling.
Dopaminergic deficits are associated with dementia in a variety of conditions (50–55). Although the positive symptoms of schizophrenia have generally been related to subcortical dopaminergic hyperactivity, many authors have suggested that in schizophrenia, some aspects of dopaminergic activity are suppressed (56–59), which could contribute to cognitive impairment (3). Low (60) and normal (61) prefrontal immunoreactivity for tyrosine hydroxylase have both been reported. If dopaminergic reserves are even focally diminished in schizophrenia, further decline with age (normally beginning in adolescence [62] and progressing through senescence [63]) could impair cognition or increase vulnerability to the effects of other aging processes.
Neuroleptic drugs could, of course, play a part. They are antagonists at a variety of receptors for dopamine, acetylcholine, serotonin, norepinephrine, and epinephrine. The authors of a report of low tyrosine hydroxylase activity in the deep layers of the anterior cingulate cortex noted the absence of this abnormality in two neuroleptic-free patients (61), raising the possibility that neuroleptic drugs influence the distribution of cortical dopamine terminals. However, the relatively low incidence of cognitive impairment in the neuroleptic-treated mood disorder patients in our study indicates that neuroleptics alone do not account for the greater sensitivity to neuritic senile plaques and neurofibrillary tangles that we find in schizophrenia.
Presented in part at the 51st annual meeting of the Society of Biological Psychiatry, New York, May 1–5, 1996; the 72nd annual meeting of the American Association of Neuropathologists, Vancouver, June 11–16, 1996; and the 26th annual meeting of the Society for Neuroscience, Washington, D.C., Nov. 16–21, 1996Received Sept. 9, 1997; ; revision received April 24, 1998; accepted May 7, 1998. From the Divisions of Neuropathology and Brain Imaging, Department of Neuroscience, and the Departments of Brain Imaging and Clinical Psychobiology, New York State Psychiatric Institute; and the Departments of Pathology, Psychiatry, Neurology, and Radiology, Columbia University, New York. Address reprint requests to Dr. Dwork, Division of Neuropathology, Unit 62, New York State Psychiatric Institute, 722 West 168th St., New York, NY 10032; [email protected] (e-mail). Supported by grants AG-10638 and AG-08702 from the National Institute on Aging and by grant MH-50727 from NIMH. Abbott Laboratories provided Alz 50 antibody.The authors thank Dr. Shu-Hui Yen for antibody to paired helical filaments.
 |
 |
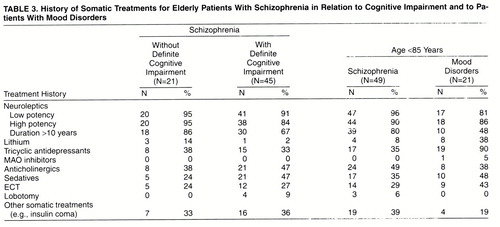 |

FIGURE 1. Mean Values for Postmortem Measures of Senile Degeneration in Elderly Psychiatric Patients and Nonpsychiatric Comparison Subjects, by Diagnosis

FIGURE 2. Mean Values for Postmortem Measures of Senile Degeneration in Elderly Schizophrenia Patients Aged <85, With and Without Definite Cognitive Impairmenta

FIGURE 3. Age-Adjusted Mean Values for Postmortem Measures of Senile Degeneration in Elderly Schizophrenia Patients Without Definite Cognitive Impairment, Aged <85, and Elderly Nonpsychiatric Subjects Without Cognitive Impairment, Aged <85a
1. Johnstone EC, Crow TJ, Frith CD, Stevens M, Krel L, Husband J: The dementia of dementia praecox. Acta Psychiatr Scand 1978; 357:305–324Crossref, Google Scholar
2. Gold JM, Weinberger DR: Cognitive deficits and the neurobiology of schizophrenia. Curr Opin Neurobiol 1995; 5:225–230Crossref, Medline, Google Scholar
3. Harvey PD, Lombardi J, Kincaid MM, Parrella M, White L, Powchik P, Davidson M: Cognitive functioning in chronically hospitalized schizophrenic patients: age-related changes and age disorientation as a predictor of impairment. Schizophr Res 1995; 17:15–24Crossref, Medline, Google Scholar
4. Harvey PD, White L, Parrella M, Putnam KM, Kincaid MM, Powchik P, Mohs RC, Davidson M: The longitudinal stability of cognitive impairment in schizophrenia: Mini-Mental State scores at one- and two-year follow-ups in geriatric in-patients. Br J Psychiatry 1995; 166:630–633Crossref, Medline, Google Scholar
5. Davidson M, Harvey PD, Powchik P, Parrella M, White L, Knobler HY, Losonczy MF, Keefe RSE, Katz S, Frecska E: Severity of symptoms in chronically institutionalized geriatric schizophrenic patients. Am J Psychiatry 1995; 152:197–207Link, Google Scholar
6. Jellinger K: Neuromorphological background of pathochemical studies in major psychoses, in Pathochemical Markers in Major Psychoses. Edited by Beckmann H, Riederer P. Berlin, Springer Verlag, 1985, pp 1–23Google Scholar
7. Buhl L, Bojsen-Møller M: Frequency of Alzheimer’s disease in a postmortem study of psychiatric patients. Dan Med Bull 1988; 35:288–290Medline, Google Scholar
8. Bruton CJ, Crow TJ, Frith CD, Johnstone EC, Owens DGC, Roberts GW: Schizophrenia and the brain: a prospective clinico-neuropathological study. Psychol Med 1990; 20:285–304Crossref, Medline, Google Scholar
9. Prohovnik I, Dwork AJ, Kaufman MA, Willson N: Alzheimer-type neuropathology in elderly schizophrenia. Schizophr Bull 1993; 19:805–816Crossref, Medline, Google Scholar
10. Purohit DP, Perl DP, Haroutunian V, Powchik P, Davidson M, Davis KL: Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry 1998; 55:205–211Crossref, Medline, Google Scholar
11. Baldessarini RJ, Hegarty JD, Bird ED, Benes FM: Meta-analysis of postmortem studies of Alzheimer’s disease-like neuropathology in schizophrenia. Am J Psychiatry 1997; 154:861–863; correction, 154:1180Link, Google Scholar
12. Gurland BJ: The borderlands of dementia: the influence of sociocultural characteristics on rates of dementia occurring in the senium, in Clinical Aspects of Alzheimer’s Disease and Senile Dementia: Aging, vol 15. Edited by Miller NE, Cohen GD. New York, Raven Press, 1981, pp 61–84Google Scholar
13. Zhang MY, Katzman R, Salmon D, Jin H, Cai G, Wang Z, Qu G, Grant I, Tu E, Levy P, Klauber MR, Liu WT: The prevalence of dementia and Alzheimer’s disease in Shanghai, China: impact of age, gender, and education. Ann Neurol 1990; 27:428–437Crossref, Medline, Google Scholar
14. Katzman R: Education and the prevalence of dementia and Alzheimer’s disease. Neurology 1993; 43:13–20Crossref, Medline, Google Scholar
15. Stern Y, Alexander GE, Prohovnik I, Stricks L, Link B, Lennon MC, Mayeux R: Relationship between lifetime occupation and parietal flow: implications for a reserve against Alzheimer’s disease pathology. Neurology 1995; 45:55–60Crossref, Medline, Google Scholar
16. Keilp JG, Waniek C, Goldman RG, Zemishlany Z, Alexander GE, Gibbon M, Wu A, Susser E, Prohovnik I: Reliability of post-mortem chart diagnoses of schizophrenia and dementia. Schizophr Res 1995; 17:221–228Crossref, Medline, Google Scholar
17. Khachaturian ZS: Diagnosis of Alzheimer’s disease. Arch Neurol 1985; 42:1097–1105Crossref, Medline, Google Scholar
18. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L: The Consortium to Establish a Registry for Alzheimer"s Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer"s disease. Neurology 1991; 41:479–486Crossref, Medline, Google Scholar
19. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK: Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 1992; 13:179–189Crossref, Medline, Google Scholar
20. Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA, Berg L: Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996; 46:707–719Crossref, Medline, Google Scholar
21. Ksiezak-Reding H, Davies P, Yen S-H: Alz 50, a monoclonal antibody to Alzheimer’s disease antigen, cross-reacts with tau proteins from bovine and normal human brain. J Biol Chem 1988; 263:7943–7947Medline, Google Scholar
22. Yen S-H, Crowe A, Dickson DW: Monoclonal antibodies to Alzheimer neurofibrillary tangles, 1: identification of polypeptides. Am J Pathol 1985; 120:282–291Medline, Google Scholar
23. Dwork AJ, Liu D, Kaufman MA, Prohovnik I: Archival, formalin-fixed tissue: its use in the study of Alzheimer’s type changes. Clin Neuropathol 1998; 17:45–49Medline, Google Scholar
24. Morris JC, McKeel DW Jr, Storandt M, Rubin EH, Price JL, Grant EA, Ball MJ, Berg L: Very mild Alzheimer’s disease: informant-based clinical, psychometric, and pathologic distinction from normal aging. Neurology 1991; 41:469–478Crossref, Medline, Google Scholar
25. Berg L, McKeel DW Jr, Miller JP, Baty J, Morris JC: Neuropathological indexes of Alzheimer’s disease in demented and nondemented persons aged 80 years and older. Arch Neurol 1993; 50:349–358Crossref, Medline, Google Scholar
26. El-Mallakh RS, Kirch DG, Shelton R, Fan K-J, Pezeshkpour G, Kanhouwa S, Wyatt RJ, Kleinman JE: The nucleus basalis of Meynert, senile plaques, and intellectual impairment in schizophrenia. J Neuropsychiatry Clin Neurosci 1991; 3:383–386Crossref, Medline, Google Scholar
27. Arnold SE, Trojanowski JQ, Gur RE, Blackwell P, Han LY, Choi C: Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry 1998; 55:225–232Crossref, Medline, Google Scholar
28. Wisniewski HM, Wen GY, Kim KS: Comparison of four staining methods on the detection of neuritic plaques. Acta Neuropathol (Berl) 1989; 78:22–27Crossref, Medline, Google Scholar
29. Harvey PD, Powchik P, Mohs RC, Davidson M: Memory functions in geriatric chronic schizophrenic patients: a neuropsychological study. J Neuropsychiatry Clin Neurosci 1995; 7:207–212Crossref, Medline, Google Scholar
30. Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A: Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988; 23:138–144Crossref, Medline, Google Scholar
31. DeKosky ST, Scheff SW: Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 1990; 27:457–464Crossref, Medline, Google Scholar
32. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30:572–580Crossref, Medline, Google Scholar
33. Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM, Kester DB, Stafiniak P: Neuropsychological function in schizophrenia: selective impairment in memory and learning. Arch Gen Psychiatry 1991; 48:618–624Crossref, Medline, Google Scholar
34. Jones P, Rodgers B, Murray R, Marmot M: Child development risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet 1994; 344:1398–1402Crossref, Medline, Google Scholar
35. Haroutunian V, Davidson M, Kanof PD, Perl DP, Powchik P, Losonczy M, McCrystal J, Purohit DP, Bierer LM, Davis KL: Cortical cholinergic markers in schizophrenia. Schizophr Res 1994; 12:137–144Crossref, Medline, Google Scholar
36. Eastwood SL, Harrison PJ: Decreased synaptophysin in the medial temporal lobe in schizophrenia demonstrated using immunoautoradiography. Neuroscience 1995; 69:339–343Crossref, Medline, Google Scholar
37. Eastwood SL, Burnet PWJ, Harrison PJ: Altered synaptophysin expression as a marker of synaptic pathology in schizophrenia. Neuroscience 1995; 66:309–319Crossref, Medline, Google Scholar
38. Browning MD, Dudek EM, Rapier JL, Leonard S, Freedman R: Significant reductions in synapsin but not synaptophysin specific activity in the brains of some schizophrenics. Biol Psychiatry 1993; 34:529–535Crossref, Medline, Google Scholar
39. Thompson PM, Sower AC, Perrone-Bizzozero MI: Altered levels of the synaptosomal associated protein SNAP 25 in schizophrenia. Biol Psychiatry 1998; 43:239–243Crossref, Medline, Google Scholar
40. Gabriel SM, Haroutunian V, Powchik P, Honer W, Davidson M, Davies P, Davis K: Increased concentrations of presynaptic proteins in the cingulate cortex of schizophrenics. Arch Gen Psychiatry 1997; 54:559–566Crossref, Medline, Google Scholar
41. Honer WG, Falkai P, Young C, Wang T, Xie J, Bonner J, Hu L, Boulianne GL, Luo Z, Trimble WS: Cingulate cortex synaptic terminal proteins and neural cell adhesion molecule in schizophrenia. Neuroscience 1997; 78:99–110Crossref, Medline, Google Scholar
42. Young CE, Arima K, Xie J, Hu L, Beach TG, Falkai P, Honer WG: SNAP-25 deficit and hippocampal connectivity in schizophrenia. Cerebral Cortex 1998; 8:261–268Crossref, Medline, Google Scholar
43. Arnold SE, Lee VA-Y, Gur RE, Trojanowski JQ: Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc Natl Acad Sci USA 1991; 88:10850–10854Crossref, Medline, Google Scholar
44. Cotter D, Kerwin R, Doshi B, Martin CS, Everall IP: Alterations in hippocampal non-phosphorylated MAP2 protein expression in schizophrenia. Brain Res 1997; 765:238–246Crossref, Medline, Google Scholar
45. Dwork AJ, Rosoklija G: Loss of subicular map2 immunoreactivity in schizophrenia is not associated with gliosis. Abstracts of the Society for Neuroscience 1997; 23:2201Google Scholar
46. Arnold SE, Franz BR, Gur RC, Gur RE, Shapiro RM, Moberg PJ, Trojanowski JQ: Smaller neuron size in schizophrenia in hippocampal subfields that mediate cortical-hippocampal interactions. Am J Psychiatry 1995; 152:738–748Link, Google Scholar
47. Benes FM, Sorensen I, Bird ED: Reduced neuronal size in posterior hippocampus of schizophrenic patients. Schizophr Bull 1991; 17:597–608Crossref, Medline, Google Scholar
48. Rajkowska G, Selemon LD, Goldman-Rakic PS: Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry 1998; 55:215–224Crossref, Medline, Google Scholar
49. Selemon LD, Rajakowska G, Goldman-Rakic PS: Abnormally high neuron density in the schizophrenic cortex: a morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry 1995; 52:805–818Crossref, Medline, Google Scholar
50. Albert ML, Feldman RG, Willis AL: The “subcortical dementia” of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 1974; 37:121–130Crossref, Medline, Google Scholar
51. Knopman DS, Mastri AR, Frey WH II, Sung JH, Rustan T: Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology 1990; 40:251–256Crossref, Medline, Google Scholar
52. Torack RM, Morris JC: Mesolimbocortical dementia: a clinicopathologic case study of a putative disorder. Arch Neurol 1986; 43:1074–1078Crossref, Medline, Google Scholar
53. Gibb WRG, Luthert PH, Marsden CD: Corticobasal degeneration. Brain 1989; 112:1171–1192Crossref, Medline, Google Scholar
54. McKeith LG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH: Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology 1996; 47:1113–1124Crossref, Medline, Google Scholar
55. Stern Y, Tetrud JW, Martin WR, Kutner SJ, Langston JW: Cognitive change following MPTP exposure. Neurology 1990; 40:261–264Crossref, Medline, Google Scholar
56. Weinberger DR: The pathogenesis of schizophrenia: a neurodevelopmental theory, in Handbook of Schizophrenia, vol 1: The Neurology of Schizophrenia. Edited by Nasrallah HA, Weinberger DR. Amsterdam, Elsevier, 1986, pp 397–406Google Scholar
57. Grace AA: Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 1991; 41:1–24Crossref, Medline, Google Scholar
58. Goldstein M, Deutch AY: Dopaminergic mechanisms in the pathogenesis of schizophrenia. FASEB J 1992; 6:2413–2421Crossref, Medline, Google Scholar
59. Wyatt RJ, Karoum F, Casanova MF: Decreased DOPAC in the anterior cingulate cortex of individuals with schizophrenia. Biol Psychiatry 1995; 38:4–12Crossref, Medline, Google Scholar
60. Akil M, Lewis DA: Reduced dopamine innervation of the prefrontal cortex in schizophrenia. Abstracts of the Society for Neuroscience 1996; 22:1679Google Scholar
61. Benes F, Todtenkopf MS, Taylor JB: Differential distribution of tyrosine hydroxylase fibers on small and large neurons in layer II of anterior cingulate cortex of schizophrenic brain. Synapse 1997; 25:80–92Crossref, Medline, Google Scholar
62. Côté LJ, Kremzner LT: Biochemical changes in normal aging in human brain, in The Dementias. Edited by Mayeux R, Rosen WG. New York, Raven Press, 1983, pp 19–30Google Scholar
63. Mann DMA, Yates PO: Lipoprotein pigments—their relationship to aging in the human nervous system, II: the melanin content of pigmented nerve cells. Brain 1974; 97:489–498Crossref, Medline, Google Scholar

