
High Loading of Polygenic Risk for ADHD in Children With Comorbid Aggression
Abstract
Objective
Although attention deficit hyperactivity disorder (ADHD) is highly heritable, genome-wide association studies (GWAS) have not yet identified any common genetic variants that contribute to risk. There is evidence that aggression or conduct disorder in children with ADHD indexes higher genetic loading and clinical severity. The authors examine whether common genetic variants considered en masse as polygenic scores for ADHD are especially enriched in children with comorbid conduct disorder.
Method
Polygenic scores derived from an ADHD GWAS meta-analysis were calculated in an independent ADHD sample (452 case subjects, 5,081 comparison subjects). Multivariate logistic regression analyses were employed to compare polygenic scores in the ADHD and comparison groups and test for higher scores in ADHD case subjects with comorbid conduct disorder relative to comparison subjects and relative to those without comorbid conduct disorder. Association with symptom scores was tested using linear regression.
Results
Polygenic risk for ADHD, derived from the meta-analysis, was higher in the independent ADHD group than in the comparison group. Polygenic score was significantly higher in ADHD case subjects with conduct disorder relative to ADHD case subjects without conduct disorder. ADHD polygenic score showed significant association with comorbid conduct disorder symptoms. This relationship was explained by the aggression items.
Conclusions
Common genetic variation is relevant to ADHD, especially in individuals with comorbid aggression. The findings suggest that the previously published ADHD GWAS meta-analysis contains weak but true associations with common variants, support for which falls below genome-wide significance levels. The findings also highlight the fact that aggression in ADHD indexes genetic as well as clinical severity.
Attention deficit hyperactivity disorder (ADHD) is a highly heritable early-onset neurodevelopmental disorder, with marked clinical heterogeneity, affecting males more often than females (1–3). It has a complex genetic architecture, as is the case for most common psychiatric disorders. Although associated rare variants have been identified (4–7), the general view is that the most satisfactory explanatory model of inheritance is a multifactorial, polygenic liability threshold one in which the combined effects of multiple common genetic variants with environmental factors contribute to ADHD risk.
With respect to common risk variants, there are no genome-wide significant findings for ADHD (8, 9). Although it has been proposed that this simply reflects inadequate sample sizes (5, 10), others suggest that the lack of findings is a consequence of psychiatric disorders (including ADHD) being explained mainly or solely by rare high-penetrance variants (11). So far, for ADHD, rare variants in the form of copy number variants (CNVs) have been found to be associated (4–7), and studies have shown that other classes of rare mutations make some contribution to autism spectrum disorder (ASD) (12–14), another childhood-onset disorder that strongly overlaps with ADHD (15). While the issue of the relative contributions of common and rare variants is far from being empirically resolved, pathway-analytic approaches have shown that not only do both contribute to ADHD, but they tend to act on similar functional classes of genes (5, 16).
Given a contribution from both common and rare alleles (and presumably also alleles with intermediate frequencies), the multifactorial, polygenic liability threshold model predicts that forms of the disorder in groups of people less often affected (e.g., ADHD in females) or with more severe forms of the disorder should carry a greater genetic load, including greater enrichment of ADHD common risk alleles.
The presence of conduct disorder in youths with ADHD is known to index greater clinical severity (17). Twin and family studies have also shown that in children with ADHD, the presence of conduct disorder symptoms indexes higher ADHD familial and genetic loading (18–22). For example, the relative risk for ADHD in biological relatives of probands who have ADHD with comorbid conduct disorder (relative risk=9.5) is almost double that of relatives of probands with ADHD alone (relative risk=5.4) (18). These studies suggest that in ADHD, the presence of conduct disorder likely indexes greater genetic load. Previously, it has only been possible to infer this indirectly by measuring recurrence rates in various classes of relatives, but recent developments now allow the component attributable to relatively common alleles to be estimated using genome-wide molecular genetic data in the form of polygenic load (23–25). In the present study, we used ADHD polygenic risk scores derived from the largest published genome-wide association meta-analysis (8) to test in an independent sample whether ADHD accompanied by conduct disorder is characterized by greater enrichment of ADHD “risk alleles.” We also investigated the relationship between polygenic score and conduct disorder symptoms.
Method
Study Subjects
Participants for what we term in this study the “Cardiff sample” were recruited from child and adolescent mental health services or community pediatric outpatient clinics in the United Kingdom. The sample of 452 children met criteria for a lifetime DSM-III-R or DSM-IV diagnosis of ADHD, confirmed by a research diagnostic assessment (26). Children with bipolar disorder, schizophrenia, ASD, Tourette’s syndrome, an IQ <70 (assessed using the WISC-III or WISC-IV) (27, 28), epilepsy, or any other neurological or genetic disorder were excluded. Written informed consent from parents and assent or consent from children was obtained for all participants. The study protocol was approved by North-West England and Wales Multicentre Research Ethics Committees.
The comparison sample comprised 5,081 individuals from the Wellcome Trust Case Control Consortium–Phase 2.
Clinical Measures
For the Cardiff sample, ADHD diagnoses were confirmed using the Child and Adolescent Psychiatric Assessment (26), a research diagnostic interview undertaken with the child’s parents. Interviews were conducted by trained psychologists who were supervised weekly by a child psychiatrist and a psychologist. Interrater reliability for diagnosis of ADHD subtype, assessed using 60 cases, was excellent (kappa=1.0). Information on symptom pervasiveness and impairment in school was obtained using the Child ADHD Teacher Telephone Interview (29) or the DuPaul (30) or Conners teacher rating scales (31).
The Child and Adolescent Psychiatric Assessment was also used to assess comorbid psychiatric disorders. Interrater reliability for parent-rated conduct disorder symptoms was very good (intraclass correlation=0.98). Summed scores for total number of DSM-IV conduct disorder symptoms were obtained in addition to diagnoses. In line with previous studies and factor analyses (32, 33), total counts for aggressive symptoms (DSM-IV conduct disorder criteria labeled as “aggression to people and animals”) and covert symptoms (DSM-IV conduct disorder criteria labeled as “destruction of property” or “deceitfulness or theft”) were generated. (For a full list of DSM-IV aggressive and covert conduct disorder symptoms, see the data supplement that accompanies the online edition of this article.)
Genotyping
DNA was obtained through saliva and blood samples. Genotyping for cases was performed on the Illumina (San Diego) Human660W-Quad BeadChip. Genotyping for comparison subjects was performed using the Illumina Human 1.2M BeadChip. The samples, quality control assessment, and genome-wide association study results have been described in detail previously (5).
Data Used to Derive ADHD Polygenic Risk Scores
We used the international meta-analysis of ADHD versus control data, described in detail elsewhere (8). This data set contains data for 2,064 trios, 896 case subjects, 2,455 comparison individuals, and 1,206,461 single-nucleotide polymorphisms (SNPs). Case subjects were assessed with the same inclusion criteria and by methods similar to those used in the Cardiff study. A total of 54 individuals with ADHD were removed from the Cardiff sample because they had been included in the meta-analysis (8). The comparison groups did not overlap.
Polygenic Analysis
We used the analytic approach described by the International Schizophrenia Consortium (25) with the published meta-analysis of ADHD (8) as the discovery data set and Cardiff sample ADHD data as the target set. Each individual in the target set was assigned a polygenic score based on information in the discovery set. We made comparisons between ADHD case subjects (with and without conduct disorder) and comparison subjects to determine whether the ADHD-derived polygenic scores were significantly different. As comorbid conduct disorder indexes higher ADHD familial loading, our key aim was to test whether polygenic scores for ADHD were higher in ADHD case subjects with conduct disorder compared to those without.
We first selected a set of SNPs in relative linkage equilibrium in our ADHD and comparison samples using a sliding window of 200 SNPs, moving it along the genome in steps of five SNPs and dropping a SNP when the pairwise estimate of linkage disequilibrium (r2) was greater than 0.2 (PLINK command:–indep-pairwise 200 5 0.2) (34). In the discovery meta-analysis data set, we identified corresponding p values and associated alleles for the selected SNPs. Based on the findings from the International Schizophrenia Consortium (25) and Psychiatric Genetics Consortium studies (23, 24), which identified a relaxed p value <0.5 as the optimal “association” threshold in discovery samples of the size equivalent to those used here, we defined this a priori as the threshold from which to derive score alleles from our discovery sample. Alleles that are more common in the discovery cases at p<0.5 for two-tailed p values are termed the “score” alleles (34). For each individual in the target sample, we used PLINK to obtain a polygenic score that corresponds to the mean number of score alleles across the set of SNPs. We employed logistic regression analysis to compare the polygenic scores for ADHD in the target set to those in the comparison sample. To allow for population stratification in our data, we conditioned on two covariates (the first two principal components estimated from our GWAS data using the EIGENSTRAT software package, which was designed for this purpose (35, 36). We hypothesized a priori that ADHD “risk alleles” derived from the published GWAS would be enriched in our independent ADHD sample (relative to the comparison sample), and in particular in those with conduct disorder. Therefore, for each analysis, we report a one-tailed p value and pseudo-R2, the latter being a measure of the estimated variability in case-control status explained.
Using logistic regression analysis, we then compared polygenic score in those with and without a conduct disorder diagnosis from the set of ADHD cases (ADHD with conduct disorder compared with ADHD without conduct disorder).
Linear regression analyses were used to investigate whether polygenic score was significantly associated with total DSM-IV conduct disorder symptom score, as well as aggression and covert conduct disorder symptom scores. Our rationale for further dividing conduct disorder symptoms into two subgroups was based on factor analyses (33) showing that they can be split into aggressive symptoms (such as cruelty to people or animals, fighting, and stealing with confrontation of the victim) and covert symptoms (such as fire-setting, breaking into a building or car, and vandalism).
Results
A total of 452 children from the Cardiff sample met inclusion criteria and had genetic and phenotypic data available. They ranged in age from 6 to 17 years (mean=10.7 years, SD=2.8); 389 were male (86.1%) and 63 were female (13.9%). The mean full-scale IQ was 87 (SD=11.2). The gender ratio and IQ scores are typical of U.K. ADHD clinic case subjects. The mean number of ADHD symptoms was 14.68 (SD=2.87; 25th percentile ADHD score=13.00, 50th percentile=15.00, 75th percentile=17.00). Within this sample, 77 individuals (17.0%) had a diagnosis of ADHD with conduct disorder and 375 (83.0%) had no comorbid conduct disorder. The mean numbers of DSM-IV conduct disorder symptoms were 3.6 (SD=1.86) and 0.5 (SD=0.76) for the ADHD groups with and without conduct disorder, respectively. There was no association between conduct disorder scores, age, and gender. In addition, 229 (50.7%) subjects met criteria for a DSM-IV diagnosis of oppositional defiant disorder; 65 of those with a conduct disorder diagnosis (84.4%) also had a diagnosis of oppositional defiant disorder. Twenty-two individuals (4.9%) had a comorbid diagnosis of anxiety disorder, and three individuals (0.7%) had a comorbid depressive disorder.
Polygenic Scores Predicting ADHD in the Target Sample
ADHD risk as defined from weakly associated alleles (N=46,156) in the discovery GWAS was significantly higher in ADHD case subjects than in comparison subjects (p=0.01) (Table 1). Thus, as we postulated, risk for ADHD is in part attributable to common alleles tagged by the genome-wide genotyping arrays.
| Comparison Analysis (Sample 1 Versus Sample 2) | Sample sizes | z | R2 (%) | p | |
|---|---|---|---|---|---|
| Sample 1 | Sample 2 | ||||
| Total ADHD sample | Comparison group | 452 and 5,081 | 2.32 | 0.098 | 0.010 |
| ADHD with conduct disorder | Comparison group | 77 and 5,081 | 3.11 | 0.190 | 0.00095 |
| ADHD, no conduct disorder | Comparison group | 375 and 5,081 | 1.27 | 0.030 | 0.10 |
| ADHD with conduct disorder | ADHD, no conduct disorder | 77 and 375 | 2.23 | 1.1 | 0.013 |
Polygenic Score Enrichment in Those With ADHD Accompanied by Conduct Disorder
The polygenic score representing ADHD risk was significantly higher in ADHD case subjects who had a conduct disorder diagnosis than in those who did not (p=0.01). The magnitude of the effect (as defined by R2) was 1.1%, larger than that observed when comparing ADHD case subjects and comparison subjects (Table 1).
To test whether our findings could be attributable to higher ADHD total symptom counts in those with conduct disorder, we tested the association between ADHD symptom count and polygenic risk score. Within ADHD case subjects, the total number of ADHD symptoms was not significantly associated with polygenic score. As expected, total ADHD score was significantly associated with total conduct disorder score (β=0.159, t=−2.900, p=0.004).
ADHD polygenic risk scores were significantly higher in female than in male case subjects (β=−0.104, t=−2.159, p=0.031), and hence all the data were reanalyzed allowing for sex as a covariate. The results were unchanged (data not shown).
Polygenic Score Predicting Conduct Disorder Symptom Scores
Within case subjects, ADHD polygenic risk score increased with total conduct disorder score (β=0.118, t=2.530, p=0.006). At the level of individual composite phenotypes, polygenic risk score also increased with number of aggressive conduct disorder symptoms (β=0.151, t=3.152, p=0.002), but not covert conduct disorder symptoms. These associations remained significant when controlling for sex. The distribution of risk scores showed increasing polygenic score with respect to increasing total conduct disorder scores (Figure 1).
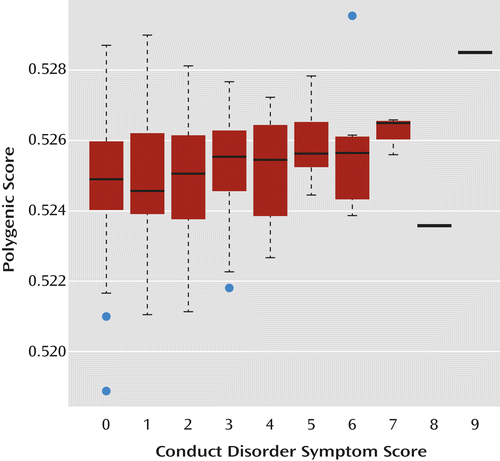
a The whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. The horizontal bars indicate median scores, and the blue dots represent outliers.
Discussion
We initiated this study to investigate the contribution of common genetic variants to ADHD and to test whether comorbid conduct disorder, defined categorically and dimensionally, indexed greater genetic risk at a molecular level. Our data support a polygenic component to ADHD, in that the risk score was higher in our independent sample of ADHD case subjects than in the comparison sample. To our knowledge, this is the first report to suggest that the previously published ADHD GWAS meta-analysis (8) harbors common risk alleles that show contribution to ADHD when they are considered en masse. More importantly, as hypothesized on the basis of previous family and twin studies suggesting that comorbid conduct disorder indexes higher familial and genetic loading in ADHD, we found that ADHD risk score is particularly elevated in those with ADHD and conduct disorder compared with those who have ADHD only.
A within-case analysis of conduct disorder symptoms as a dimension rather than a category revealed similar findings, with a positive linear relationship between ADHD polygenic scores and comorbid conduct disorder symptoms. Interestingly, this association was related to aggressive, rather than covert, conduct disorder symptoms. Twin studies also suggest that these different symptom dimensions may be distinct in their genetic etiology, with stronger genetic loading for overt aggressive symptoms (32, 37).
Overall, our study confirms the hypothesis that common genetic variants are relevant to ADHD risk. Our findings also highlight the fact that comorbid conduct disorder indexes heterogeneity in terms of genetic loading at a molecular level. Our finding that individual symptom groups of total conduct disorder scores and aggressive conduct disorder scores are significantly associated with polygenic score further underscore the point that specific clinical phenotypes can index differential genetic loading. Most of the evidence to date suggests that conduct problems index ADHD cases that are quantitatively rather than qualitatively different from the remaining ADHD cases (38) in terms of the patterns of association with clinical, cognitive, genetic, and environmental correlates. This is in keeping with the approach taken by ICD-10, in which hyperkinetic conduct disorder is considered a subtype of ADHD/hyperkinetic disorder (39). However, some associated factors also appear to be unique to conduct disorder in ADHD. Notably, the functional COMT Val158Met variant has been found to be associated with conduct problems in ADHD (40), a finding that has been replicated in six independent samples (32, 41–43), while meta-analysis shows that this variant is not associated with ADHD risk (44). A separate issue of interest would be to investigate the independent contribution of genetic liability associated with conduct disorder (regardless of ADHD). There is evidence to suggest shared liabilities, but as yet, large-scale conduct disorder GWAS data sets are unavailable.
We also incidentally found that female case subjects had significantly higher ADHD risk scores than males. This finding requires replication, but it is intriguing, as it supports the hypothesis that ADHD is less common in females because a more extreme genetic load is required for the liability threshold to be surpassed. This would predict that relatives of female ADHD probands have a greater risk for ADHD than relatives of male ADHD probands, although evidence here is lacking (3). However, there is a possible effect of ascertainment bias. Females with ADHD may be less likely to be diagnosed (or referred for diagnosis) than males with comparable severity of disorder (45), and therefore our findings might simply reflect a higher threshold of severity for females to be diagnosed and ascertained. However, females in our sample did not have significantly more ADHD or conduct disorder symptoms than males.
As expected, ADHD severity (i.e., total ADHD score) showed association with conduct disorder scores, but the genetic findings were driven by conduct disorder, as there was no association between ADHD severity and polygenic risk. Given that all clinical case subjects have high ADHD scores by definition, and thus variance is limited, this may not be surprising.
It is noteworthy that case-control comparisons were significant for the total ADHD group and for the ADHD with conduct disorder group but did not achieve significance in the ADHD without conduct disorder group. This result might simply reflect sample size, whereby larger samples would be needed to demonstrate statistically significant differences in groups with lower genetic load (those with ADHD without conduct disorder) than in those with higher genetic load (those with ADHD with conduct disorder).
This study used a well-characterized sample of children who underwent careful phenotyping. The diagnosis of ADHD was confirmed using a semistructured research diagnostic interview, which also allowed the collection of detailed information on conduct disorder symptoms, and the measures showed high reliability. It is not possible to obtain clinical data on the comparison subjects, although failure to exclude comparison subjects with ADHD or conduct disorder would reduce our study’s statistical power, not generate false positives. One limitation of case-control and within-case studies is the possibility of population stratification. However, we limited the impact of stratification by the well-accepted approach of including derived principal components that allow for population variation (35, 36) in our ADHD GWAS and by using hypothesis-driven analyses. Furthermore, for stratification to be an issue, it would have to be refractory to inclusion of the principal components, and the same uncorrected ethnic variation would have to be overrepresented in case subjects in both the Cardiff sample and the samples used in the meta-analysis (46); it would also have to be specifically overrepresented in subjects with comorbid conduct disorder compared with the full ADHD sample.
The magnitudes of the polygenic effect (as defined by R2) are small, as is typical when this method is used. The magnitudes are also smaller than some of those published for schizophrenia (R2=3%–6% [24, 25]) but not all (47). This method is also very sensitive to sample size, and available GWAS data sets for ADHD are very much smaller than those for schizophrenia. In the most recent schizophrenia analysis, with more than 9,000 case subjects and 12,000 comparison individuals (24), the total amount of variance explained was estimated to be 6%, whereas in an earlier analysis of 3,000 case subjects and 3,000 comparison subjects by the International Schizophrenia Consortium (25), the estimate was 3%. Our estimate that 0.1% of the variance explained is based on much smaller samples, with 452 ADHD case subjects and 5,081 comparison subjects. Another factor that would reduce explained variance is heterogeneity across samples. This could plausibly be higher for some disorders than others—for example, through international differences in clinical service provision and thus in ascertainment, as well as other variability, such as ethnic composition. It is also important to note that GWAS SNP arrays do not completely capture relevant genetic variation; they “tag” potentially causal variants, and the arrays do not capture the full spectrum of allelic frequencies or allele types (e.g., repeat sequence polymorphisms such as variable number tandem repeats). Overall, we expect that as more ADHD and comparison samples with more comprehensive genetic capture become available, more variance will be explained. Finally, we also acknowledge the contribution of environmental risk factors and gene-environment interplay, although this was not the focus of the present analyses.
These findings suggest that ADHD, like other psychiatric disorders, can be considered a polygenic disorder, the architecture of which includes common as well as rare alleles (10). They are also compatible with our earlier work showing overlap between the biological processes enriched for weak ADHD SNP association signals and those enriched for rare copy number variants (5). Given the evidence for contribution of common variants, the future acquisition and genetic analysis of much larger ADHD samples in an attempt to capture relevant genetic variation is crucial. The aim is not to simply identify single “significant” SNPs of small effect size, but rather to utilize the spectrum of associated common and rare genetic risk variants to uncover novel clues for risk mechanisms and underlying biology and to inform our conceptualization of ADHD.
In summary, we found that common genetic variation appears relevant to ADHD and that a higher loading for common ADHD genetic risk variants is indexed by comorbid conduct disorder, especially aggressive symptoms. Our results also suggest that the previously published ADHD GWAS meta-analysis contains weak but true associations to common variants, support for which falls below currently accepted genome-wide significance levels. The findings highlight the fact that aggression in ADHD, as an index of clinical severity, is underpinned by higher genetic loading at a molecular level. They also illustrate that for hypothesis-driven research, careful phenotyping is still useful in psychiatric genetic studies.
1 : Genetic basis of attention deficit and hyperactivity. Br J Psychiatry 1999; 174:105–111Crossref, Medline, Google Scholar
2 : Molecular genetics of attention deficit hyperactivity disorder. Psychiatr Clin North Am 2010; 33:159–180Crossref, Medline, Google Scholar
3 : Attention-deficit disorder and conduct disorder in girls: evidence for a familial subtype. Biol Psychiatry 2000; 48:21–29Crossref, Medline, Google Scholar
4 : Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am J Psychiatry 2012; 169:195–204Link, Google Scholar
5 ;
6 : Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376:1401–1408Crossref, Medline, Google Scholar
7 : Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry 2011; 16:491–503Crossref, Medline, Google Scholar
8 ;
9 : Genome-wide association studies in ADHD. Hum Genet 2009; 126:13–50Crossref, Medline, Google Scholar
10 : Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13:537–551Crossref, Medline, Google Scholar
11 : Genomic analysis of mental illness: a changing landscape. JAMA 2010; 303:2523–2524Crossref, Medline, Google Scholar
12 : Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012; 485:242–245Crossref, Medline, Google Scholar
13 : De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485:237–241Crossref, Medline, Google Scholar
14 : Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012; 485:246–250Crossref, Medline, Google Scholar
15 : Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur Child Adolesc Psychiatry 2010; 19:281–295Crossref, Medline, Google Scholar
16 : Integrated genome-wide association study findings: identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am J Psychiatry 2011; 168:365–377Link, Google Scholar
17 : Hyperactivity and conduct problems as risk factors for adolescent development. J Am Acad Child Adolesc Psychiatry 1996; 35:1213–1226Crossref, Medline, Google Scholar
18 : Toward guidelines for pedigree selection in genetic studies of attention deficit hyperactivity disorder. Genet Epidemiol 2000; 18:1–16Crossref, Medline, Google Scholar
19 : Examining the comorbidity of ADHD-related behaviours and conduct problems using a twin study design. Br J Psychiatry 2001; 179:224–229Crossref, Medline, Google Scholar
20 : Genetic and environmental influences on the covariation between hyperactivity and conduct disturbance in juvenile twins. J Child Psychol Psychiatry 1996; 37:803–816Crossref, Medline, Google Scholar
21 : Test of alternative hypotheses explaining the comorbidity between attention-deficit/hyperactivity disorder and conduct disorder. J Abnorm Child Psychol 2008; 36:29–40Crossref, Medline, Google Scholar
22 : Phenotypic and measurement influences on heritability estimates in childhood ADHD. Eur Child Adolesc Psychiatry 2010; 19:311–323Crossref, Medline, Google Scholar
23 ;
24 ;
25 ;
26 : The Child and Adolescent Psychiatric Assessment (CAPA). Psychol Med 1995; 25:739–753Crossref, Medline, Google Scholar
27 : Wechsler Intelligence Scale for Children, 3rd ed. San Antonio, Tex, Psychological Corp, 1992Google Scholar
28 : Wechsler Intelligence Scale for Children, 4th ed (WISC-IV): Administration and Scoring Manual. San Antonio, Tex, Psychological Corp, 2003Google Scholar
29 : The Child Attention-Deficit Hyperactivity Disorder Teacher Telephone Interview (CHATTI): reliability and validity. Br J Psychiatry 2004; 184:74–78Crossref, Medline, Google Scholar
30 : Parent and teacher ratings of ADHD symptoms: psychometric properties in a community-based sample. J Clin Child Psychol 1991; 20:245–253Crossref, Google Scholar
31 : A teacher rating scale for use in drug studies with children. Am J Psychiatry 1969; 126:884–888Link, Google Scholar
32 : Genetic risk for conduct disorder symptom subtypes in an ADHD sample: specificity to aggressive symptoms. J Am Acad Child Adolesc Psychiatry 2009; 48:757–764Crossref, Medline, Google Scholar
33 : Oppositional defiant disorder and conduct disorder: a meta-analytic review of factor analyses and cross-validation in a clinic sample. Clin Psychol Rev 1993; 13:319–340Crossref, Google Scholar
34 : PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–575Crossref, Medline, Google Scholar
35 : Population structure and eigenanalysis. PLoS Genet 2006; 2:e190Crossref, Medline, Google Scholar
36 : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38:904–909Crossref, Medline, Google Scholar
37 : Do aggressive and non-aggressive antisocial behaviors in adolescents result from the same genetic and environmental effects? Am J Med Genet B Neuropsychiatr Genet 2004; 129B:59–63Crossref, Medline, Google Scholar
38 : Autism symptoms in attention-deficit/hyperactivity disorder: a familial trait which correlates with conduct, oppositional defiant, language, and motor disorders. J Autism Dev Disord 2009; 39:197–209Crossref, Medline, Google Scholar
39 : Co-transmission of conduct problems with attention-deficit/hyperactivity disorder: familial evidence for a distinct disorder. J Neural Transm 2008; 115:163–175Crossref, Medline, Google Scholar
40 : Catechol O-methyltransferase gene variant and birth weight predict early-onset antisocial behavior in children with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 2005; 62:1275–1278Crossref, Medline, Google Scholar
41 : A replicated molecular genetic basis for subtyping antisocial behavior in children with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 2008; 65:203–210Crossref, Medline, Google Scholar
42 : Variation in the catechol-O-methyltransferase Val 158 Met polymorphism associated with conduct disorder and ADHD symptoms, among adolescent male delinquents. Psychiatr Genet 2010; 20:20–24Crossref, Medline, Google Scholar
43 : Genotype link with extreme antisocial behavior: the contribution of cognitive pathways. Arch Gen Psychiatry 2010; 67:1317–1323Crossref, Medline, Google Scholar
44 : Candidate gene studies of ADHD: a meta-analytic review. Hum Genet 2009; 126:51–90Crossref, Medline, Google Scholar
45 : Child and family factors influencing the clinical referral of children with hyperactivity: a research note. J Child Psychol Psychiatry 1997; 38:479–485Crossref, Medline, Google Scholar
46 : Polygenic dissection of the bipolar phenotype. Br J Psychiatry 2011; 198:284–288Crossref, Medline, Google Scholar
47 ;

