
The Evolution of the Cognitive Model of Depression and Its Neurobiological Correlates
Abstract
Although the cognitive model of depression has evolved appreciably since its first formulation over 40 years ago, the potential interaction of genetic, neurochemical, and cognitive factors has only recently been demonstrated. Combining findings from behavioral genetics and cognitive neuroscience with the accumulated research on the cognitive model opens new opportunities for integrated research. Drawing on advances in cognitive, personality, and social psychology as well as clinical observations, expansions of the original cognitive model have incorporated in successive stages automatic thoughts, cognitive distortions, dysfunctional beliefs, and information-processing biases. The developmental model identified early traumatic experiences and the formation of dysfunctional beliefs as predisposing events and congruent stressors in later life as precipitating factors. It is now possible to sketch out possible genetic and neurochemical pathways that interact with or are parallel to cognitive variables. A hypersensitive amygdala is associated with both a genetic polymorphism and a pattern of negative cognitive biases and dysfunctional beliefs, all of which constitute risk factors for depression. Further, the combination of a hyperactive amygdala and hypoactive prefrontal regions is associated with diminished cognitive appraisal and the occurrence of depression. Genetic polymorphisms also are involved in the overreaction to the stress and the hypercortisolemia in the development of depression—probably mediated by cognitive distortions. I suggest that comprehensive study of the psychological as well as biological correlates of depression can provide a new understanding of this debilitating disorder.
I was privileged to start my research on depression at a time when the modern era of systematic clinical and biological research was just getting underway. Consequently, the field for new investigations was wide open. The climate at the time was friendly for such research. The National Institute of Mental Health had only recently been funding research and providing salary support for full-time clinical investigators. The Group for Advancement of Psychiatry, under the leadership of pioneers like David Hamburg, was providing guidelines as well as the impetus for clinical research.
Caught up with the contagion of the times, I was prompted to start something on my own. I was particularly intrigued by the paradox of depression. This disorder appeared to violate the time-honored canons of human nature: the self-preservation instinct, the maternal instinct, the sexual instinct, and the pleasure principle. All of these normal human yearnings were dulled or reversed. Even vital biological functions like eating or sleeping were attenuated. The leading causal theory of depression at the time was the notion of inverted hostility. This seemed a reasonable, logical explanation if translated into a need to suffer. The need to punish one’s self could account for the loss of pleasure, loss of libido, self-criticism, and suicidal wishes and would be triggered by guilt. I was drawn to conducting clinical research in depression because the field was wide open—and besides, I had a testable hypothesis.
Cross-Sectional Model of Depression
I decided at first to make a foray into the “deepest” level: the dreams of depressed patients. I expected to find signs of more hostility in the dream content of depressed patients than nondepressed patients, but they actually showed less hostility. I did observe, however, that the dreams of depressed patients contained the themes of loss, defeat, rejection, and abandonment, and the dreamer was represented as defective or diseased. At first I assumed the idea that the negative themes in the dream content expressed the need to punish one’s self (or “masochism”), but I was soon disabused of this notion. When encouraged to express hostility, my patients became more, not less, depressed. Further, in experiments, they reacted positively to success experiences and positive reinforcement when the “masochism” hypothesis predicted the opposite (summarized in Beck [1] ).
Some revealing observations helped to provide the basis for the subsequent cognitive model of depression. I noted that the dream content contained the same themes as the patients’ conscious cognitions—their negative self-evaluations, expectancies, and memories—but in an exaggerated, more dramatic form. The depressive cognitions contained errors or distortions in the interpretations (or misinterpretations) of experience. What finally clinched the new model (for me) was our research finding that when the patients reappraised and corrected their misinterpretations, their depression started to lift and—in 10 or 12 sessions—would remit (2) .
Thus, I undertook the challenge of attempting to integrate the different psychological pieces of the puzzle of depression. The end product was a comprehensive cognitive model of depression. At the surface, readily accessible level was the negativity in the patients’ self-reports, including their dreams and their negative interpretations of their experiences. These variables seemed to account for the manifestations of depression, such as hopelessness, loss of motivation, self-criticism, and suicidal wishes. The next level appeared to be a systematic cognitive bias in information processing leading to selective attention to negative aspects of experiences, negative interpretations, and blocking of positive events and memories. These findings raised the question: “What is producing the negative bias?” On the basis of clinical observations supported by research, I concluded that when depressed, patients had highly charged dysfunctional attitudes or beliefs about themselves that hijacked the information processing and produced the negative cognitive bias, which led to the symptoms of depression (1 , 3 , 4) .
A large number of studies have demonstrated that depressed patients have dysfunctional attitudes, show a systematic negative attentional and recall bias in laboratory experiments, and report cognitive distortions (selective abstraction, overgeneralizing, personalization, and interpretational biases [4] ). Dysfunctional attitudes, measured by the Dysfunctional Attitudes Scale (5) , are represented by beliefs such as “If I fail at something, it means I’m a total failure.” During a full-blown episode of depression, the hypersalient dysfunctional attitudes lead into absolute negative beliefs about the self, their personal world, and the future (“I am a failure”). I suggested that these dysfunctional attitudes are embedded within cognitive structures, or schemas, in the meaning assignment system and thus have structural qualities, such as stability and density as well as thresholds and levels of activation. The degree of salience (or “energy”) of the schemas depends on the intensity of a negative experience and the threshold for activation at a given time (successive stressful experiences, for example, can lower the threshold [1] ).
When the schemas are activated by an event or series of events, they skew the information processing system, which then directs attentional resources to negative stimuli and translates a specific experience into a distorted negative interpretation. The hypersalience of these negative schemas leads not only to a global negative perception of reality but also to the other symptoms of depression, such as sadness, hopelessness, loss of motivation, and regressive behaviors such as social withdrawal and inactivity. These symptoms are also subjected to negative evaluation (“My poor functioning is a burden on my family” and “My loss of motivation shows how lazy I am”). Thus, the depressive constellation consists of a continuous feedback loop with negative interpretations and attentional biases with the subjective and behavioral symptoms reinforcing each other.
Developmental Model of Depression
Cognitive Vulnerability
What developmental event or events might lead to the formation of dysfunctional attitudes and how these events might relate to later stressful events leading to the precipitation of depression was another piece of the puzzle. In our earlier studies, we found that severely depressed patients were more likely than moderately or mildly depressed patients to have experienced parental loss in childhood (6) . We speculated that such a loss would sensitize an individual to a significant loss at a later time in adolescence or adulthood, thus precipitating depression. Brij Sethi, a member of our group, showed that the combination of a loss in childhood with an analogous loss in adulthood led to depression in a significant number of depressed patients (7) . The meaning of the early events (such as “If I lose an important person, I am helpless”) is transformed into a durable attitude, which may be activated by a similar experience at a later time. A recent prospective study observed that early life stress sensitizes individuals to later negative events through impact on cognitive vulnerability leading to depression (8) .
The accumulated research findings have supported the original cognitive vulnerability model derived from clinical observations (4) . As shown in Figure 1 , early adverse events foster negative attitudes and biases about the self, which are integrated into the cognitive organization in the form of schemas; the schemas become activated by later adverse events impinging on the specific cognitive vulnerability and lead to the systematic negative bias at the core of depression (1 , 9) .
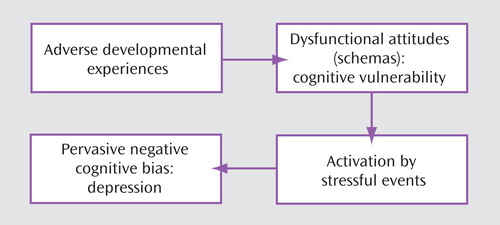
Much of the early research by others on the cognitive model overlooked the role of stress in activating previously latent dysfunctional schemas. Scher and colleagues (10) provided a comprehensive review of the diathesis-stress formulation based on prospective studies and priming methodologies to test the cognitive model.
Although the original cognitive model proposed that severe life events (e.g., death of a loved one or loss of a job) were the usual precipitants of depression (1) , more recent research has suggested that milder stressful life events provide an alternate pathway to depression in vulnerable individuals (11 , 12) . Moreover, the triggering events of successive episodes of depression become progressively milder, suggesting a “kindling” effect (13 , 14) .
Cognitive vulnerability to the experience of depressive symptoms following stress has been reported in children, adolescents, and adults (10 , 12 , 15) . For example, students showing cognitive vulnerability were more likely to become depressed following negative outcomes on college applications than were students not showing cognitive vulnerability (12 , 16) . It should be noted that these studies generally described minor depressive episodes rather than full-blown major depression.
What relevance do dysfunctional attitudes have to the vulnerability to recurrence of depression? Segal and colleagues (17) showed that the muted dysfunctional attitudes of recovered depressed patients could be primed by a negative mood induction procedure. Furthermore, the extent to which the mood induction activated the dysfunctional attitudes during the nondepressed period predicted future relapse and recurrence. This activation of the dysfunctional attitudes was more likely to occur with patients who had received pharmacotherapy than those receiving cognitive therapy. The prospective and priming studies thus indicated that dysfunctional attitudes could be regarded as a cognitive vulnerability factor for depression.
A more recent refinement of the cognitive vulnerability model has added the concept of cognitive reactivity , expressed clinically as fluctuations in patients’ negative attitudes about themselves in response to daily events (18) . Cognitive reactivity has been demonstrated experimentally by a variety of priming interventions (or “mood interventions”) such as sad music, imaging of sad autobiographical memories, social rejection film clip, or contrived failure. Following these priming interventions, clinically vulnerable subjects report more dysfunctional attitudes, negative cognitive biases, and erosion of normal positive biases than do other subjects (10) . Clinical vulnerability was defined in terms of high-risk variables (e.g., a personal or family history of depression).
Studies have shown that the presence of cognitive reactivity before a stressful life event predicts the onset of depressive episodes (10) . The importance of the meaning assigned to a stressful event as a crucial component of cognitive reactivity was borne out by the finding (19) that the daily negative appraisals of daily stressors predicted daily depressive symptoms. The addition of the concept of cognitive reactivity to the cognitive model suggested that the predisposition to depression may be observable in the daily cognitive-emotional reactions of the depression-prone individual (18) . However, the model left unanswered why certain individuals were more reactive to daily events (or were more likely to develop dysfunctional attitudes and cognitive biases) than others.
As indicated previously, the experience of episodes of depressive symptoms is different from the total immersion of the personality in a full-blown major depression. Severe depression is characterized not only by a broad range of intense symptoms but also “endogenous” features, such as relative insensitivity to external events. To account for the complex characteristics of the fully expressed depression, I proposed an expanded cognitive model (4 , 20) . I presented the concept of the mode , a network of cognitive, affective, motivational, behavioral, and physiological schemas, to account for the profound retardation, anhedonia, and sleep and appetite disturbance, as well as the cognitive aberrations. The activation of this mode (network) produces the various phenomena of depression.
In the formation of the mode, the connections among the various negatively oriented schemas become strengthened over time in response to negatively interpreted events. Successive symptom-producing events or a major depressogenic event locks these connections into place. In a sense, the cognitive schemas serve as the hub and the other schemas as nodes with continuous communication among them. A major stressful event or events symbolizing a loss of some type trigger the cognitive schemas that activate the other (affective, motivational, etc.) schemas. When fully activated, the mode becomes relatively autonomous and is no longer as reactive to external stimuli; that is, positive events do not reduce the negative thinking or mood. Attentional resources are disproportionately allocated from the external environment to internal experiences such as negative cognitions and sadness, manifested clinically as rumination. Also, resources are withdrawn from adaptive schemas such as coping and problem solving. The mode presumably would correspond to a complex neural network, including multiple relevant brain regions that are activated or deactivated during depression.
The negatively biased cognitive schemas function as automatic information processors. The biased automatic processing is rapid, involuntary, and sparing of resources. The dominance of this system (efficient but maladaptive) in depression could account for the negative attentional and interpretational bias. In contrast, the role of the cognitive control system (consisting of executive functions, problem solving, and reappraisal) is attenuated during depression. The operation of this system is deliberate, reflective, and effortful (resource demanding), can be reactivated in therapy, and, thus, can be used to appraise the depressive misinterpretations and dampen the salience of the depressive mode.
The concept of the two forms of processing can be traced back to Freud’s model of primary and secondary processes (21) and has been reformulated many times since (22) . Recently, Beevers (23) suggested a similar formulation of a two-factor processing in depression. He proposed that cognitive vulnerability to depression occurs when negatively biased associative processing is uncorrected by reflective processing.
The expanded cognitive model includes the following progression in the development of depression: adverse early life experiences contribute to the formation of dysfunctional attitudes incorporated within cognitive structures, labeled cognitive schemas (cognitive vulnerability). When activated by daily life events, the schemas produce an attentional bias, negatively biased interpretations, and mild depressive symptoms (cognitive reactivity).
After repeated activation, the negative schemas become organized into a depressive mode, which also includes affective, behavioral, and motivational schemas (cognitive vulnerability). Accumulated negative events or a severe adverse event impacts on the mode and makes it hypersalient. The hypersalient mode takes control of the information processing, reflected by increased negative appraisals and rumination. The cognitive control of emotionally significant appraisals is attenuated and, thus, reappraisal of negative interpretations is limited. The culmination of these processes is clinical depression. Crick and Dodge (24) point out that with repeated activation, maladaptive information-processing patterns become routinized and resistant to change. Thus, cognitive schemas, after repeated activation before and during depressive episodes, become more salient and more ingrained over time, consistent with the “kindling” phenomenon.
Although supportive evidence for this model was meager in early years, support for the fundamental hypotheses has accumulated over the past 40 years. In 1999, David A. Clark and I (4) reviewed over 1,000 publications relevant to the cognitive model of depression and found substantial research support for the various facets of the cognitive theory. Several more recent reviews have provided additional research support (10 , 12 , 15 , 25) .
As the cognitive model of depression buttressed by years of systematic research has grown to maturity, it seems timely and appropriate to compare it with the burgeoning findings in neurogenetics and neuroimaging.
Biological Correlates of the Cognitive Model
Genetic Vulnerability
I still had an unsolved problem: how can we account for the observation that only a proportion of individuals subjected to child abuse, other adverse events, and major traumatic experiences become depressed? Many of us had speculated about the existence of a “blue gene,” but the technology for identifying it had not been available. Also, although there was considerable support for the cognitive model of depression at the psychological and clinical levels (4) , there were minimal data from neurophysiological studies to correlate with these findings. It was not until this century that these problems could be addressed, as a result of investigations by researchers in behavioral genetics and cognitive neuroscience. The spectacular technological advances in genetics and functional neuroimaging have enabled researchers to demonstrate that genetic variations and their impact on neural functioning play a major role in the hyperreactivity to negative experiences leading to depression. A number of studies have provided a structure for understanding the relationships between life events, neural dysregulation, cognitive processes, and depression. This research has also provided a preliminary basis for formulating the neurobiological correlates of such psychological constructs as cognitive vulnerability, cognitive reactivity, and cognitive biases.
These advances have facilitated a breakthrough in understanding the relationship between cognitive, biological, and experiential factors in the development of depression. The genetic and neurobiological findings illuminated some probable causal pathways to depression as well as suggesting biological correlates of the cognitive model. The pioneering paper by Caspi and colleagues (26) suggested that individuals possessing either one or two copies of the short variant of the 5-HTTLPR (serotonin transporter) gene, which is not transcriptionally as effective as the long form, experienced higher levels of depression and suicidality following a recent life stressor. The study by Caspi and colleagues (26) has since been supported by a large number of other studies (27) .
The types of stressful life events moderated by the 5-HTTLPR gene varied considerably in these studies, ranging from mild stressors to a single, large traumatic event. Also, in some studies, adverse experiences, for example, abuse in childhood (28) , appear to represent a distal predisposition to depression, whereas in others, 5-HTTLPR genotype moderates the depressogenic effects of more proximal events (11) . Studies of both predispositional (biological and psychological) and precipitating events would provide a test of this aspect of the cognitive model. Kilpatrick and colleagues (29) found that carriers of the low-expression variant of the short-form 5-HTTLPR polymorphism were prone to develop major depression and postdisaster posttraumatic stress disorder under conditions of high exposure to hurricanes and low social support. Kaufman and colleagues (28) have also found evidence that social support buffers against depressive reactions to stressful experiences among genetically vulnerable individuals. Although questions have been raised regarding the generalizability of the 5-HTTLPR findings (e.g., 30 ), they are useful to illustrate parallels between biological and psychological concepts. The more exhaustive analyses would include other variants such as the CREB and COMT genes (31 , 32) .
Investigators have also found that the brain-derived neurotrophic factor genotype interacted with the 5-HTTLPR gene to predict depression in children (28) and older adults (33) . Of interest, variants of the brain-derived neurotrophic factor predicted ruminations and depression differently in adolescent girls and their mothers (34) . Specifically, Hilt and colleagues (34) found that girls with the Val/Val genotype had higher rumination scores and exhibited more symptoms of depression than girls with the Val/Met genotype. In contrast, mothers with adult -onset depression and the Val/Met genotype exhibited more symptoms of depression and rumination. Of interest, mothers with childhood -onset depression were more likely to have the Val/Val genotype. Kaufman and colleagues (28) found that in maltreated children, depressive severity was predicted in part by an interaction of the 5-HTTLPR (short allele) with the brain-derived neurotrophic factor ( Val/Met ) genotype, particularly among children with low social support. A variant of the HTR-2A gene has been found to potentiate the effect of maternal nurturance in mitigating the experience of depressive symptoms in children (35) . As research proceeds in this area, it seems likely that a variety of other gene-gene and gene-environment interactions will be discovered. In general, the established relationships between environmental events, biological predisposition, and depression appear to run parallel to the findings of the cognitive model regarding environmental events, cognitive factors, and depression.
Genetic Diathesis and Cognitive Bias
While the genetic studies have pointed to the innate biological vulnerability to stress leading to depression, the relation of biological to cognitive vulnerability needed to be clarified. The gene-by-environment findings for depression have prompted an interest in uncovering their relationships to cognitive variables. A variety of experimental procedures have been used to test for cognitive bias in individuals at genetic risk for depression. The negative attentional, recall, and interpretative biases are generally elicited by mood induction procedures, such as viewing sad movie clips or imagining sad experiences (36) . Genetic antecedents for these observed cognitive biases have been identified. A number of studies indicate that negative cognitive processing and negative cognitions are associated with the presence of the 5-HTTLPR short allele (37 – 40) . Of particular relevance to possible cognitive predisposition, Hayden and colleagues (40) found that nondepressed children homozygous for the short allele showed greater negative processing on a self-referential encoding task following a negative mood induction than did children with other genotypes. Thus, the accumulating evidence suggests the genetic predisposition to depression is associated with biases in the processing of information.
Neurophysiological and Cognitive Factors/Bias
What neurophysiological processes are related to the cognitive biases? Multiple findings have tied amygdala hyperactivity to depression (41 , 42) . However, the findings need to be considered within a broader framework, including many brain regions implicated in depression (43 , 44) . A specific line of inquiry has tied the 5-HTTLPR variant to activation in brain regions critical for processing negative stimuli. Hyperreactivity of the amygdala in the short 5-HTTLPR variant carriers is associated with increased sensitivity to negative stimuli (45) and leads to negative bias in the processing or interpretation of emotional stimuli (46 , 47) . Since the amygdala is involved in the evaluation and storage of emotionally charged events (48) , its hyperreactivity to negative stimuli in predisposed individuals would appear to represent a neurophysiological correlate of cognitive bias.
The systematic bias in information processing in depression is reflected not only in selective attention and exaggerated reaction to negative stimuli (49 , 50) but also in the expectancy of aversive events (51) . Abler and colleagues (51) found that the anticipation of noxious stimuli produced excessive amygdala activation in genetically prone individuals. Further, there is evidence that the 5-HTTLPR gene interacted with children’s attentional and inferential biases to predict depressive symptoms; inferential bias alone predicts lifetime diagnosis of depression among carriers of the 5-HTTLPR short allele, but not among those homozygous for the long allele (unpublished work by Gibb BE, Benas JS, Grassia M, McGeary J and unpublished work by Gibb BE, Uhrlass DJ, Grassia M, McGeary J).
The pathway from genetic and cognitive predisposition to depression may be clarified by studies of the impact of stress on neural functioning. Gotlib and colleagues (52) have found that carriers of the short-form serotonin transporter gene (5-HTTLPR) show elevated cortisol response, cognitive biases, and activation of the amygdala during a mood repair procedure. Adverse circumstances engage the hypothalamic-pituitary-adrenal (HPA) axis, which leads to the secretion of excessive “stress hormones” such as cortisol (52) . Presumably, the continual secretion of cortisol culminates in the hypercortisolemia characteristic of most depressed individuals (1 , 53) .
Several converging findings suggest that the cognitive appraisal of a stressor plays a role in the evocation of cortisol response and the generation of depressive symptoms. In a review of relevant literature, Dickerson and Kemeny (54) , for example, noted consistent findings that experimental manipulations appraised as threat of social rejection produced an elevated cortisol response. These results, combined with the findings by Hankin and colleagues (19) on the negative cognitive responses to daily stressful events leading to depressive symptoms, suggest a pathway to depressive symptoms: stress→ distorted appraisal→ engagement of the HPA axis→ cortisol→ depressive symptoms. Of course, there are undoubtedly feedback loops involving both psychological and biological variables.
A further elaboration of this hypothesis is suggested by Gotlib (unpublished work by Gotlib IH), who proposed a reciprocal model involving the dysregulated HPA axis (in response to specific stressors) leading to increased cortisol secretion, which affects the serotonergic system. He also proposes a more complex theory that involves the impact of increased cortisol secretion on the short form of the 5-HTTLPR gene, leading to an alteration in the transmission of serotonin and, consequently, negative feedback to the HPA axis and increased cortisol secretion.
Gotlib’s theory can be amplified to take into account research findings demonstrating interactions between measures of cognitive reactivity and serotonin. Studies have shown that increases or decreases in serotonin activity are related to self-assessment of dysfunctional attitudes or cognitive reactivity. Meyer and colleagues (55) reported that acute tryptophan depletion of serotonin increases dysfunctional attitudes, while Booij and colleagues (56) found that depletion of serotonin increased cognitive reactivity. The finding that individuals with the short variant of the 5-HTTLPR gene show depressive symptoms after experimental depletion of serotonin provides evidence of the association of genomic with neurochemical vulnerability. Finally, Meyer and colleagues (57) reported increased dysfunctional attitudes in depressed patients with low intracellular serotonin. Thus, the preliminary evidence suggests linkages between cognitive vulnerability and genetic vulnerability expressed as a hyperreactive serotonergic system.
Recent neurophysiological research is pertinent to another aspect of the expanded cognitive model, mainly the formulation that during depression of the cognitive control system, top-down processing is dysregulated while the bottom-up schematic processing is prepotent. Siegle and colleagues (41) found that nearly all depressed patients have reduced prefrontal function and about one-half have increased amygdala activity. Thus, the balance of their respective activity is relevant to cognitive control. Banks and colleagues (58) found that the reduced amygdala coupling with the orbitofrontal cortex and the dorsal medial prefrontal cortex predicts the extent of attenuation of negative affect following reappraisal. Johnstone and colleagues (59) point out that a key feature underlying the pathophysiology of major depression is the dysfunctional engagement of the right prefrontal cortex and the lack of engagement of the left lateral ventral medial prefrontal circuitry, important for the down-regulation of amygdala responses to negative stimuli. They suggest that the top-down process of reappraisal is defective in depressed individuals; this may account for the importance of reappraisal in the cognitive therapy of this disorder. Thus, two concurrent processes are involved in emotional processing in depression: diminished cognitive control from prefrontal and cingulate regions and increased activity in the amygdala and other regions.
Deconstructing Depression
Interpretation of the research comparing components of the cognitive model with the neurophysiological investigations of depression poses a philosophical problem. How can one reconcile two totally different levels of abstraction: mentalism and materialism? The cognitive and neurophysiological approaches use different concepts, research strategies, and technical procedures. Given this philosophical problem, is there justification for mixing the two models in terms of causation or interaction (for example, reduction of serotonin causes an increase in dysfunctional attitudes; see reference 57 ) or are the neurophysiological and cognitive processes simply “different sides of the same coin,” as I once argued (60) ? According to my earlier notion, the cognitive processes are parallel to but do not interact with the biological processes.
Notwithstanding this philosophical problem, I believe that it is possible to present a pragmatic formulation of the interaction of the two levels ( Figure 2 ). In deconstructing the phenomenon of depression, I propose a hypothetical pathway starting with genetic vulnerability (including predispositional but not protective genes). The 5-HTTLPR polymorphism leads to excessive reactivity of the amygdala (45) . The heightened limbic reactivity to emotionally significant events triggers deployment of increased attentional resources to such events, manifested by negative attentional bias and recall (cognitive reactivity). The selective focus on the negative aspects of experience results in the familiar cognitive distortions such as exaggeration, personalization, and overgeneralization (6) and, consequently, in the formation of dysfunctional attitudes regarding personal adequacy, acceptability, and worth. Frequent reiterations of negative interpretations shape the content of the cognitive schemas (unlovable, inadequate, worthless). Concurrently, the negative interpretations of experience have an impact on the HPA axis and set in motion the previously described cycle involving the overreactive serotonergic system and consequently lead to depression. Care needs to be taken in the interpretation of gene-environment studies, including those of the 5-HTTLPR variant, given the numerous possible methodological pitfalls in these sorts of analyses (Kenneth Kendler, personal communication, May 11, 2008). Consequently, this particular formulation is tentative, subject to future research.
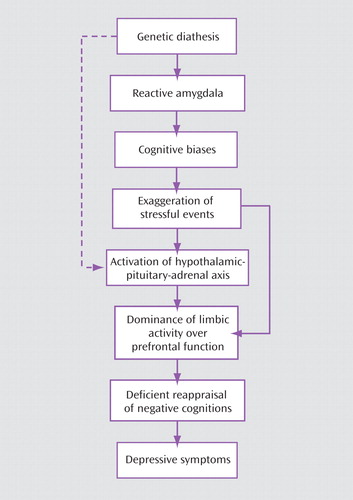
a Multiple interactions are not shown. Genetic pathways leading to reduced prefrontal activity have not been determined as yet. Increased limbic activity overrides prefrontal control.
The theory of the consolidation of the negative attitudes and the core negative self-concept along with the associated affective, motivational, and behavioral factors into the depressive mode is speculative but is useful as an explanatory construct (20) . The progression from depressive proneness to a full-blown depression would involve not only the hyperactivation of this conglomerate but also the diminution of reality testing. The biological counterpart of this theoretical model includes complex circuits involving multiple brain regions. Mayberg (61) , for example, defines a major depressive episode as a “pattern of dysfunctional interactions among specific cingulate, paralimbic, subcortical, and frontal regions critical to maintaining emotional homeostasis under conditions of exogenous or endogenous stress” (p. 258). At present the most direct parallel involves the predominance of negatively biased processing and reduced reality testing on the one hand and amygdala activation and disengagement of executive (especially prefrontal) regions on the other.
Future Perspectives
The accumulation of studies of the psychological and biological aspects of depression has reached a critical mass warranting a new synthesis. The findings of relationships among the diverse genetic, neurophysiological, environmental, and cognitive aspects of the vulnerability to and development of depression call for future studies integrating these findings into a comprehensible formulation. A series of multiple wave prospective studies could identify the relevant variables and their interrelationships. The studies ideally would be complementary to each other so that each finding would contribute to the overall formulation of the theory of depression. The clarification of specific associations among the relevant variables should yield valuable information. In order to simplify the suggested plan, I have limited the genetic diathesis to the 5-HTTLPR gene. Obviously, other genes, including those that have a protective effect, as well as the individuals’ social support should be included in any formulations. Moreover, as indicated previously, the research based on this gene is not totally reliable.
The proposed psychobiological model of depression poses a number of problems and questions that need to be addressed. The developmental model presupposes that the 5-HTTLPR gene in some as yet unidentified way leads to hyperreactivity of the amygdala to external stimuli. This activity is associated with a negative cognitive bias. In addition, increased reactivity of the HPA axis is also associated with this genetic variant (52) . A longitudinal study starting in childhood could investigate the causal sequence. In one pathway, the hyperactive amygdala leads initially to attentional bias that gradually progresses into significant negative cognitive distortions of daily experiences that engage the HPA axis. Alternatively, the amygdala and HPA overactivity may contribute independently to the cognitive and physiological impact of life events.
A number of other problems warrant further investigation. The association of increased dysfunctional attitudes with serotonin depletion poses an interesting question: is the association due to a common linkage to some undefined third factor, such as amygdala reactivity? Does serotonin deficiency cause an increase in dysfunctional attitudes (55) , is the converse true, or is there another explanation for this association? The finding that depressed individuals treated with cognitive therapy, compared to pharmacotherapy treated patients, do not show an increase in depressive symptoms (generally associated with dysfunctional attitudes) following tryptophan depletion calls for further investigation (62) . These results indicate a more complex relationship between serotonin depletion and dysfunctional attitudes. The neurobiological mechanisms involved in the reduced reality testing of negative beliefs deserve further attention. The specific brain areas involved in dysconnectivity need to be spelled out. Is the preemption of amygdala activity over prefrontal and cingular executive functions related to reduction of serotonin inhibition of the amygdala (63) ? Work relating poor error detection in depression to specific dysfunctions in various brain regions (64) needs to be followed.
The integrative developmental model postulates that various genetic variants sensitize individuals to life experiences that make them vulnerable to depression. Specifically, the studies would address the biological mechanisms that contribute to depression through the tendency to construe events in an excessively negative way. A series of waves, starting in early childhood, should examine the variables associated with the polymorphism: assessments of information processing biases (36 , 37 , 40) , negative cognitions (38) , and dysfunctional attitudes (65) following negative mood inductions. These findings would then be compared with studies of brain activity to determine their associations with the limbic system as well as prefrontal cingular and other regions. The early studies would determine whether automatic cognitive processing precedes the development of negative cognitions and dysfunctional attitudes. Another wave could examine diary records of daily dysfunctional cognitions in response to stressful situations and relate these to cortisol responses to specific stimuli situations. Overall, these assessments over a long time span would integrate findings from neuroimaging and neuroendocrine responses to stressors with cognitive responses to daily stressful activities as well as to major life events. The association of increased dysfunctional attitudes with serotonin depletion would also be determined in these studies, and in addition the attempt to find the specific relationships among these variables could be determined.
I have reason to hope that future research will perhaps provide a new paradigm which for the first time can integrate findings from psychological and biological studies to build a new understanding of depression.
1. Beck AT: Depression: Clinical, Experimental, and Theoretical Aspects. New York Harper & Row, 1967.Google Scholar
2. Rush AJ, Beck AT, Kovacs M, Hollon SD: Comparative efficacy of cognitive therapy and pharmacotherapy in the treatment of depressed outpatients. Cognit Ther Res 1977; 1:7–37Google Scholar
3. Beck AT: Cognitive models of depression. J Cogn Psychother: An Int Quarterly 1987; 1:5–37Google Scholar
4. Clark DA, Beck AT: Scientific Foundations of Cognitive Theory and Therapy of Depression. New York, John Wiley & Sons, 1999Google Scholar
5. Beck AT, Steer RA, Brown GK, Weissman A: Factor analysis of the Dsyfunctional Attitude Scale in a clinical population. Psychological Assessment 1991; 3:478–483Google Scholar
6. Beck AT, Sethi B, Tuthill R: Childhood bereavement and adult depression. Arch Gen Psychiatry 1963; 9:295–302Google Scholar
7. Sethi BB: The relationship of separation to depression. Arch Gen Psychiatry 1964; 10:486–496Google Scholar
8. Harkness KL, Lumley MN: Child abuse and neglect and the development of depression in children and adolescents, in Handbook of Depression in Children and Adolescents. Edited by Abela JR, Hankin BL. New York, Guilford, 2008, pp 466–488Google Scholar
9. Beck AT: Cognitive Therapy and the Emotional Disorders. New York, Meridian, 1976Google Scholar
10. Scher C, Ingram R, Segal Z: Cognitive reactivity and vulnerability: empirical evaluation of construct activation and cognitive diatheses in unipolar depression. Clin Psychol Rev 2005; 25:487–510Google Scholar
11. Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B: The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry 2005; 62:529–535Google Scholar
12. Abela JRZ, Hankin BL: Cognitive vulnerability to depression in children and adolescents: a developmental psychopathology perspective, in Handbook of Depression in Children and Adolescents. Edited by Abela JRZ, Hankin BL. New York, Guilford, 2008, pp 35–78Google Scholar
13. Kendler KS, Thornton LM, Gardner CO: Stressful life events and previous episodes in the etiology of major depression in women: an evaluation of the “kindling” hypothesis. Am J Psychiatry 2000; 157:1243–1251Google Scholar
14. Monroe SM, Harkness KL: Life stress, the “kindling” hypothesis, and the recurrence of depression: considerations from a life stress perspective. Psychol Rev 2005; 112:417–445Google Scholar
15. Jacobs RH, Reinecke MA, Gollan JK, Kane P: Empirical evidence of cognitive vulnerability for depression among children and adolescents: a cognitive science and developmental perspective. Clin Psychol Rev 2008; 28:759–728Google Scholar
16. Abela JRZ, D’Alessandro DU: Beck’s cognitive theory of depression: a test of the diathesis-stress and causal mediation components. Br J Clin Psychol 2002; 41:111–128Google Scholar
17. Segal ZV, Gemar M, Williams S: Differential cognitive response to a mood challenge following successful cognitive therapy or pharmacotherapy for unipolar depression. J Abnorm Psychol 1999; 108:3–10Google Scholar
18. Butler AC, Hokanson JE, Flynn HA: A comparison of self-esteem lability and low trait self-esteem as vulnerability factors for depression. J Pers Soc Psychol 1994; 66:166–177Google Scholar
19. Hankin BL, Fraley C, Abela JRZ: Daily depression and cognitions about stress: evidence for a trait-like depressogenic cognitive style and the prediction of depressive symptoms in a prospective daily diary study. J Pers Soc Psychol 2005; 88:673–685Google Scholar
20. Beck AT: Beyond belief: a theory of modes, personality, and psychopathology, in Frontiers of Cognitive Therapy. Edited by Salkovskis P. New York, Guilford, 1996, pp 1–25Google Scholar
21. Freud S: Collected Works of Sigmund Freud. New York and Washington, DC, BiblioBazaar, originally published in 1920Google Scholar
22. Chaiken S, Trope Y: Dual-Process Theories in Social Psychology. New York, Guilford, 1999Google Scholar
23. Beevers CG: Cognitive vulnerability to depression: a dual process model. Clin Psychol Rev 2005; 25:975–1002Google Scholar
24. Crick NR, Dodge KA: A review and reformulation of social information-processing mechanisms in children’s social adjustment. Psychol Bull 1994; 115:74–101Google Scholar
25. Dozois DJA, Beck AT: Cognitive schemas, beliefs and assumptions, in Risk Factors for Depression. Edited by Dobson KS, Dozois DJA. Oxford, UK, Elsevier (in press)Google Scholar
26. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington HL: Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386–389Google Scholar
27. Uher R, McGuffin P: The moderation by the serotonin transporter gene of environmental adversity in the aetiology of mental illness: review and methodological analysis. Molecular Psychiatry 2008; 13:131–146Google Scholar
28. Kaufman J, Yang B, Douglas-Palumberi H, Grasso D, Lipschitz D, Houshyar S: Brain-derived neurotrophic factor 5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry 2006; 59:673–680Google Scholar
29. Kilpatrick DG, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Resnick HS: The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry 2007; 164:1693–1699Google Scholar
30. Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG: The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med 2005; 35:101–111Google Scholar
31. Zubenko GS, Hughes HB III: Effects of the G(-656), a variant on CREB1 promoter activity in a neuronal cell line: interactions with gonadal steroids and stress. Mol Psychiatry 2008 (Epub ahead of print)Google Scholar
32. Wichers M, Aguilera M, Kenis G, Krabbendam L, Myin-Germeys I, Jacobs N, Peeters F, Derom C, Vlietinck R, Mengelers R, Delespaul P, van Os J: The catechol-O-methyl transferase Val(158)Met polymorphism and experience of reward in the flow of daily life. Neuropsychopharmacology 2007 (Epub ahead of print)Google Scholar
33. Kim J, Stewart R, Kim S, Yang S, Shin I, Kim Y: Interactions between life stressors and susceptibility genes (5-HTTPR and BDNF) on depression in Korean elders. Biol Psychiatry 2007; 62:423–428Google Scholar
34. Hilt LM, Sander LC, Nolen-Hoeksema S, Simen AA: The BDNF Val66Met polymorphism predicts rumination and depression differently in young adolescent girls and their mothers. Neurosci Lett 2007; 429:12–16Google Scholar
35. Jokela M, Keltikangas-Järvinen L, Kivimäki M, Puttonen S, Elovainio M, Rontu R: Serotonin receptor 2A gene and the influence of childhood maternal nurturance on adulthood depressive symptoms. Arch Gen Psychiatry 2007; 64:356–360Google Scholar
36. Joormann J, Talbot L, Gotlib IH: Biased processing of emotional information in girls at risk for depression. J Abnorm Psychol 2007; 116:135–143Google Scholar
37. Beevers CG, Gibb BE, McGeary JE, Miller IW: Serotonin transporter genetic variation and biased attention for emotional word stimuli among psychiatric inpatients. J Abnorm Psychol 2007; 116:208–212Google Scholar
38. Beevers CG, Scott WD, McGeary C, McGeary JE: Negative cognitive response to a sad mood induction: associations with polymorphisms of the serotonin transporter (5-HTTLPR) gene. Cognition and Emotion (in press)Google Scholar
39. Canli T, Lesch K: Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci 2007; 10:1103–1109Google Scholar
40. Hayden EP, Dougherty LR, Maloney B, Olino TM, Durbin CE, Sheihk HI: Early-emerging cognitive vulnerability to depression and the serotonin transporter promoter region polymorphism. J Affect Disord 2008; 107:227–230Google Scholar
41. Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME: Increased amygdala and decreased dorsolateral prefrontal bold responses in unipolar depression: related and independent features. Biol Psychiatry 2007; 61:198–209Google Scholar
42. Surguladze S, Brammer MJ, Keedwell P, Giampietro V, Young AW, Travis MJ: A differential pattern of neural response toward sad versus happy facial expressions in major depressive disorder. Biol Psychiatry 2005; 57:201–209Google Scholar
43. Mayberg HS: Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull 2003; 65:193–207Google Scholar
44. Phillips ML, Drevets WC, Rauch SL, Lane R: Neurobiology of emotion perception II: implications for major psychiatric disorders. Biol Psychiatry 2003; 54:515–528Google Scholar
45. Munafò M, Brown S, Hariri A: Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol Psychiatry 2008; 63:852–857Google Scholar
46. Dannlowski U, Ohrmann P, Bauer J, Kugel H, Arolt V, Heindel W: Amygdala reactivity to masked negative faces is associated with automatic judgmental bias in major depression: a 3T fMRI study. J Psychiatry Neurosci 2007; 32:423–429Google Scholar
47. Monk CS, Klein RG, Telzer EH, Schroth EA, Mannuzza S, Moulton JL III, Guardino M, Masten CL, McClure-Tone EB, Fromm S, Blair RJ, Pine DS, Ernst M: Amygdala and nucleus accumbens activation to emotional facial expressions in children and adolescents at risk for major depression. Am J Psychiatry 2008; 165:90–98Google Scholar
48. LeDoux JE: The Emotional Brain: The Mysterious Underpinnings of Emotional Life. New York, Simon & Schuster, 1996Google Scholar
49. Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS: Can’t shake that feeling: fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol Psychiatry 2002; 51:693–707Google Scholar
50. Siegle GJ, Carter CS, Thase ME: Use of fMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am J Psychiatry 2006; 163:735–738Google Scholar
51. Abler B, Erk S, Herwig U, Walter H: Anticipation of aversive stimuli activates extended amygdala in unipolar depression. J Psychiatr Res 2007; 41:511–522Google Scholar
52. Gotlib IH, Joormann J, Minor K, Hallmayer J: HPA axis reactivity: A mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol Psychiatry 2008; 63:847–851Google Scholar
53. Parker KJ, Schatzberg AF, Lyons DM: Neuroendocrine aspects of hypercortisolism in major depression. Hormones & Behavior 2003; 43:60–66Google Scholar
54. Dickerson SS, Kemeny ME: Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol Bull 2004; 130:355–391Google Scholar
55. Meyer JH, McMain S, Kennedy SH, Korman L, Brown GM, DaSilva JN, Wilson AA, Blak T, Eynan-Harvey R, Goulding VS, Houle S, Links P: Dysfunctional attitudes and 5-HT 2 receptors during depression and self-harm. Am J Psychiatry 2003; 160:90–99 Google Scholar
56. Booij L, Van der Does AJW: Cognitive and serotonergic vulnerability to depression: convergent findings. J Abnorm Psychol 2007; 116:86–94Google Scholar
57. Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N: Brain serotonin transporter binding potential measured with carbon 11–labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry 2004; 61:1271–1279Google Scholar
58. Banks SJ, Eddy KT, Angstadt M, Nathan PJ, Phan KL: Amygdala-frontal connectivity during emotion regulation. Soc Cogn Affect Neurosc 2007; 2:303–312Google Scholar
59. Johnstone T, van Reekum CM, Urry HL, Kalin NH, Davidson RJ: Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosc 2007; 27:8877–8884Google Scholar
60. Beck AT: Cognitive therapy, behavior therapy, psychoanalysis, and pharmacotherapy: a cognitive continuum, in Psychotherapy Research: Where Are We and Where Should We Go? Edited by Williams JBW, Spitzer RL. New York, Guilford, 1984, pp 114–135Google Scholar
61. Mayberg HS: Defining neurocircuits in depression: insights from functional neuroimaging studies of diverse treatments. Psychiatr Ann 2006; 4:258–267Google Scholar
62. O’Reardon JP, Chopra MP, Bergan A, Gallop R, DeRubeis RJ, Crits-Christoph P: Response to tryptophan depletion in major depression treated with either cognitive therapy or selective serotonin reuptake inhibitor antidepressants. Biol Psychiatry 2004; 55:957–959Google Scholar
63. Stutzmann GE, McEwen BS, LeDoux JE: Serotonin modulation of sensory inputs to the lateral amygdala: dependency on corticosterone. J Neurosci 1998; 18:9529–9538Google Scholar
64. Holmes AJ, Pizzagalli DA: Spatiotemporal dynamics of error processing dysfunctions in major depressive disorder. Arch Gen Psychiatry 2008; 65:179–188Google Scholar
65. Abela JRZ, Skitch SA: Dysfunctional attitudes, self-esteem, and hassles: cognitive vulnerability to depression in children of affectively ill parents. Behav Res Ther 2007; 45:1127–1140Google Scholar

