
The ACTH Response to Dexamethasone in PTSD
Abstract
OBJECTIVE: Enhanced negative feedback and reduced adrenal output are two different models that have been put forth to explain the paradoxical observations of increased release of corticotropin-releasing factor in the face of low cortisol levels in posttraumatic stress disorder (PTSD). To discriminate between these models, the authors measured levels of adrenocorticopic hormone (ACTH) and cortisol at baseline and in response to dexamethasone in medically healthy subjects with and without PTSD. Under conditions of enhanced negative feedback inhibition, ACTH levels would not be altered relative to cortisol levels, but the ACTH response to dexamethasone would be augmented, in concert with the enhanced cortisol response to dexamethasone. In contrast, under conditions of reduced adrenal output, ACTH levels would be expected to be higher at baseline relative to cortisol levels, but the ACTH response to dexamethasone would be unchanged in PTSD relative to healthy comparison subjects. METHOD: The ACTH and cortisol responses to 0.50 mg of dexamethasone were assessed in 19 subjects (15 men and four women) with PTSD and 19 subjects (14 men and five women) without psychiatric disorder. RESULTS: The ACTH-to-cortisol ratio did not differ between groups before or after dexamethasone, but the subjects with PTSD showed greater suppression of ACTH (as well as cortisol) in response to dexamethasone. CONCLUSIONS: The data support the hypothesis of enhanced cortisol negative feedback inhibition of ACTH secretion at the level of the pituitary in PTSD. Pituitary glucocorticoid receptor binding, rather than low adrenal output, is implicated as a likely mechanism for this effect.
We have previously posited that posttraumatic stress disorder (PTSD) is characterized by an inhibition of the hypothalamic-pituitary-adrenal (HPA) axis through enhanced negative feedback. This hypothesis was initially generated by the finding of an exaggerated cortisol suppression in PTSD patients following the administration of a low dose of dexamethasone and of a greater dexamethasone-induced decline in cytosolic lymphocyte glucocorticoid receptors in combat veterans with PTSD than in those without PTSD (1). The interpretation of greater suppression of cortisol in response to dexamethasone as evidence for enhanced negative feedback rested on the assumption that the dexamethasone suppression test (DST) provides a sensitive test of the negative feedback effects of glucocorticoids (2), particularly at the level of the pituitary (3, 4).
The model of enhanced negative feedback inhibition at the level of the pituitary is compatible with a number of observations pertaining to the HPA axis in individuals with PTSD, including high levels of corticotropin-releasing factor (CRF) in CSF (5), blunted adrenocorticotropic hormone (ACTH) responses to corticotropin-releasing hormone (CRH) (6), and enhanced ACTH responses to doses of metyrapone high enough to completely suppress cortisol production (7) and cortisol-mediated negative feedback inhibition, particularly insofar as these abnormalities occur in the presence of low or normal cortisol levels (8).
Several findings in the neuroendocrinology of PTSD, however, are not compatible with enhanced negative feedback inhibition. These include a greater ACTH response to CRF in PTSD than in comparison subjects (9) and a greater ACTH response to current psychosocial stress among women with histories of childhood physical and sexual abuse than among women without such histories (10). These findings, coupled with observations of low cortisol levels at baseline and after dexamethasone administration, have given rise to an alternative hypothesis, that PTSD may be characterized by subclinical adrenal insufficiency or low adrenal output (11).
Although both enhanced negative feedback and low adrenal output can account for many of the HPA findings that have been observed in PTSD, particularly for low or normal cortisol levels in the face of high CRF levels, these two explanations would have different predictions regarding ACTH levels in PTSD (12). Negative feedback acts at the level of the pituitary (as well as at other sites in the HPA axis). Therefore, enhanced negative feedback inhibition through increased glucocorticoid receptor sensitivity should inhibit ACTH levels and cortisol levels to similar degrees, since, according to this model, cortisol production by the adrenal glands is primarily dependent on ACTH release. In contrast, low adrenal output is likely to result in a higher ratio of ACTH to cortisol, which would be particularly marked if hypothalamic CRF levels were high. That is, enhanced negative feedback would not alter the ACTH-to-cortisol ratio, whereas low adrenal output should result in an increased ACTH-to-cortisol ratio.
In this study we examined ACTH and its relationship to cortisol in response to the administration of a low dose (0.50 mg) of dexamethasone. Since the ACTH response to dexamethasone is thought to reflect an action of dexamethasone at the level of the pituitary, particularly at low blood levels of dexamethasone (3, 4), the assessment of ACTH following dexamethasone administration provides a more direct measure of feedback inhibition than does the level of circulating cortisol. Under conditions of enhanced negative feedback inhibition, the suppressive effect of dexamethasone on ACTH would be as strong as its effect on cortisol, if not stronger, because of the proposed greater sensitivity of pituitary glucocorticoid receptors. However, under conditions of low adrenal output, the effect of dexamethasone on ACTH should be normal since abnormalities in HPA axis functioning are occurring downstream of the pituitary gland.
Method
Participants
Twenty-nine men and nine women age 23 to 79 years (mean=54.47 years, SD=17.27) participated in the study. Recruitment was through advertisements placed in local newspapers or on bulletin boards around the Mount Sinai Hospital and the Bronx Veterans Affairs (VA) Medical Center requesting volunteers (including comparison subjects) for one of several research projects examining hormonal responses following extremely stressful life events. All procedures were approved by the institutional review boards at the Mount Sinai School of Medicine and the Bronx VA Medical Center, and all subjects provided written informed consent before their participation. Data on cortisol and glucocorticoid receptors from 16 of the current subjects (10 with PTSD, six without PTSD) have been previously reported (13).
Subjects were included in the study if they had an axis I diagnosis of PTSD or if they were free from a current or past history of PTSD or any other axis I disorder. Subjects with PTSD were excluded if they had comorbid substance abuse or dependence, bipolar disorder, psychosis, or obsessive-compulsive disorder but not if they were suffering from a unipolar mood disorder or other anxiety disorder (i.e., major depressive disorder, dysthymia, generalized anxiety disorder, panic disorder, social phobia, or agoraphobia). Gulf War veterans and adult children of Holocaust survivors were recruited for study, but their data were not included in the current analyses, as they will be presented elsewhere. Thirty subjects were excluded because of having had a major medical, endocrinological, or neurological illness and/or receiving standing doses of psychotropic agents or other medications likely to affect the HPA axis. Of the 38 subjects included, 21 subjects were not taking any medications. None of the remaining 17 subjects was receiving standing doses of psychotropic medications, but three were taking as-needed sedatives for sleep (i.e., zolpidem or trazodone). Examples of the other common medications that were not exclusionary were lipid-lowering agents, histamine-2 blockers, and nonsteroidal anti-inflammatory agents. Subjects were not withdrawn from medications for the purpose of study participation. As the study was conducted primarily at a VA medical center, the study group was predominantly male and does not reflect the typical predominance of women in epidemiologic samples of PTSD patients.
Clinical Assessments
Information about lifetime traumatic life events and the ages at which these events occurred was obtained by using the Trauma History Questionnaire (14). The most disturbing experience reported by an individual was defined as the “focal” event and formed the basis of an interview for PTSD using the Clinician-Administered PTSD Scale (15). At this time, events were reviewed to determine whether they met the DSM-IV criterion A involving both 1) potential life threat or threat to physical integrity and 2) subjective distress (e.g., fear, helplessness, horror). In the group with PTSD, the focal events were combat-related events involving witnessing death and mutilation or experiencing mortal danger (N=10), traumatic separation from parents in early childhood (N=2), witnessing mass execution (N=1), torture (N=1), gunshot wound (N=1), rape in adulthood (N=1), attempted strangulation (N=1), domestic violence (N=1), and childhood physical abuse (N=1). Fifteen of the 19 subjects without PTSD reported experiencing potentially traumatic events, as evidenced by positive responses on one or more items of the Trauma History Questionnaire (e.g., mugging, assault, motor vehicle accident), and six of these reported events qualified as criterion A traumatic experiences (i.e., serious motor vehicle accident, combat, intruder with weapon in home, boat accident, near drowning in friend’s pool, and losing a friend in the World Trade Center bombing on 9/11). However, no subject endorsed current or lifetime symptoms sufficient or severe enough to qualify for any psychiatric diagnosis, including adjustment disorder or acute stress disorder. Thus, even with the inclusion of the subjects exposed to traumatic events, this comparison group constituted a homogenous group of symptom-free individuals.
Other axis I diagnoses were made by using the Structured Clinical Interview for DSM-IV (16). The diagnostic interviews were conducted by a trained psychologist (S.L.H.) or psychiatrist (J.A.G., L.M.B.) with established interrater reliability, and the diagnoses were reviewed at a consensus conference by clinicians blind to the biological data.
Biological Measures
Baseline blood samples were obtained at 8:00 a.m. before dexamethasone administration for the determination of cortisol and ACTH levels. The participants ingested an oral dose of 0.5 mg of dexamethasone at 11:00 p.m., and blood samples for determination of cortisol, ACTH, and dexamethasone levels were obtained at 8:00 a.m. on the day following dexamethasone ingestion.
Plasma cortisol levels were determined by radioimmunoassay, as previously described (17). The intra-assay and interassay coefficients of variation were 4.0% and 6.8%, respectively. ACTH levels were assayed by using a commercially available kit (IncStar, Stillwater, Minn.). The intra-assay and interassay coefficients of variation were 4.7% and 7.1%, respectively. Dexamethasone levels were measured by using commercially available antibodies (IgG Corp., Nashville, Tenn.) according to a previously published method (18). The intra- and interassay coefficients of variation for dexamethasone were 8.0% and 9.0%, respectively.
The results of the DST for ACTH and cortisol are expressed as both postdexamethasone hormonal levels and percentage suppression, i.e., 100 × [(predexamethasone hormone – postdexamethasone hormone) / predexamethasone hormone].
Data Presentation and Statistical Analyses
Group differences in cortisol and ACTH levels and in ACTH-to-cortisol ratios were examined by using analysis of variance (ANOVA) or covariance (ANCOVA). For the primary analyses comparing the hormonal responses to dexamethasone of the two groups, repeated-measures ANCOVA was used with the within-subjects factor being day (i.e., pre- or postdexamethasone). The hormone values were substantially kurtotic for predexamethasone cortisol (kurtosis=4.90) and for postdexamethasone ACTH (kurtosis=6.27); the former kurtosis likely reflects an extremely low predexamethasone cortisol level in the non-PTSD group, and the latter reflects a floor effect in response to dexamethasone administration. Therefore, the natural logarithms of both the pre- and postdexamethasone hormone values were used in all analyses, with the exception of calculations of percent suppression of ACTH and cortisol following dexamethasone administration. The raw data, however, will also be reported so that they can be compared to other observations in the literature.
The difference of two logarithms is equivalent to the logarithm of their ratio. Therefore, testing the effect of day (pre- versus postdexamethasone) in the repeated-measures analysis is equivalent to examining whether the log-transformed ratio of pre- to postdexamethasone ACTH or cortisol levels is significantly different from zero and, accordingly, provides the relative change in ACTH and cortisol values, respectively, from pre- to postdexamethasone. Analyses of the interactions of day with between-subjects variables are equivalent to tests of group differences in the respective log-transformed pre- to postdexamethasone ratios (i.e., they represent group differences in the relative change, respectively, in ACTH and cortisol following dexamethasone). Tests of the interactions of day with covariates in the analyses are equivalent to regression analyses of the logarithms of ratios.
Before analyses for the purpose of hypothesis testing were performed, initial analyses to identify potential confounds in the data were undertaken. Age, ethnicity, gender, medication status, weight, height, body mass index, diagnosis of major depression, and plasma dexamethasone level were tested individually for association with both the raw and log-transformed biological outcome measures. This screening process revealed a significant gender effect for the predexamethasone ACTH level (F=10.28, df=1, 36, p=0.003) and for both the raw (F=4.41, df=1, 36, p=0.05) and log-transformed (F=14.62, df=1, 36, p=0.001) postdexamethasone ACTH levels, justifying the use of gender as a covariate in analyses of ACTH and of the ACTH-to-cortisol ratio. None of the other variables assessed showed a significant association with the biological variables. For medication status in particular, receiving any medication at all was not significantly associated with the raw or log-transformed cortisol or ACTH level or with the ACTH-to-cortisol ratio, either at baseline or after dexamethasone administration (for all analyses, F≤1.69, df=1, 36, p≥0.20). The use of sedatives was similarly not significantly associated with any of the aforementioned hormonal values (in all cases, F≤0.68, df=1, 36, p≥0.41). A diagnosis of major depression was not significantly associated with any of the preceding variables for the group as a whole (in all cases, F≤2.30, df=1, 36, p≥0.14) or for the subgroup with PTSD (in all cases, F≤2.86, df=1, 17, p>0.10). Thus, neither major depressive disorder nor medication status was used as a covariate.
Lastly, the plasma dexamethasone level was not found to correlate significantly with either the raw or log-transformed value of ACTH (respectively, r=–0.16, p=0.34, and r=–0.22, p=0.19, covaried for gender, df=35) or cortisol (r=–0.01, p=0.98, and r=–0.07, p=0.69, N=38). However, the plasma concentration of dexamethasone differed significantly between the subjects with and without PTSD (F=4.87, df=1, 36, p=0.04) (Table 1); therefore, dexamethasone level was used as a covariate in all analyses involving postdexamethasone hormone values. Pearson’s correlations and partial correlations controlling for gender and, where appropriate, for dexamethasone level were performed to assess relationships among the biological measures and between these measures and symptom severity assessments.
Results
Characteristics of the Study Group
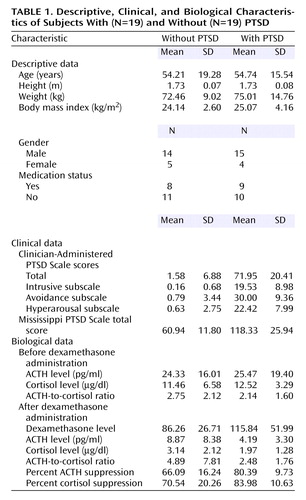
Demographic characteristics of the subjects are reported in Table 1. Nineteen participants met the diagnostic criteria for PTSD. Eleven of those meeting criteria for PTSD also had a comorbid diagnosis of major depressive disorder (N=9), dysthymia (N=2), or another anxiety disorder (N=5). The ethnic groups included Caucasian (N=23), African American (N=10), Hispanic (N=3), and Asian (N=2). The participants in the groups with and without PTSD were comparable in age (F=0.01, df=1, 36, n.s.), gender (χ2=0.15, df=1, n.s.), ethnic distribution (Caucasian versus other, χ2=2.75, df=1, n.s.), and medication status (χ2=0.11, df=1, n.s.) (Table 1). Not surprisingly, there were significant group differences in PTSD symptom severity as measured by the intrusive (F=87.92, df=1, 36, p<0.0005), avoidance (F=162.92, df=1, 36, p<0.0005), and hyperarousal (F=126.36, df=1, 36, p<0.0005) subscales of the Clinician-Administered PTSD Scale as well as the total scale (F=202.73, df=1, 36, p<0.0005) and the Mississippi PTSD Scale (F=69.52, df=1, 33, p<0.0005).
ACTH and Cortisol Suppression in PTSD
ACTH responses to dexamethasone are illustrated in Figure 1, grouped by presence or absence of PTSD diagnosis; group means are presented in Table 1. Repeated-measures ANCOVA of pre- and postdexamethasone ACTH levels revealed a significant group-by-day interaction (F=13.17, df=1, 34, p=0.001), demonstrating a greater reduction of ACTH following dexamethasone ingestion in individuals with than without PTSD. In this within-subject analysis, neither gender nor plasma dexamethasone level was a significant covariate. When the results were expressed as the percentage of suppression of ACTH, one-way ANCOVA demonstrated a significant main effect of group (F=12.59, df=1, 34, p=0.001), reflecting significantly greater dexamethasone-induced suppression of ACTH in the PTSD group. Again, neither gender nor dexamethasone level was a significant covariate.
With respect to cortisol, repeated-measures ANCOVA similarly demonstrated a significant group-by-day interaction (F=7.29, df=1, 35, p=0.02), indicating a greater reduction in cortisol following dexamethasone ingestion in individuals with than without PTSD. Plasma dexamethasone level was not a significant covariate in the interaction with day. When these relationships were expressed as the percentage of cortisol suppression, one-way ANCOVA demonstrated a significant main effect of group (F=5.08, df=1, 35, p=0.04), without a significant effect of plasma dexamethasone level.
In order to estimate adrenal responsiveness in PTSD, we examined the ACTH-to-cortisol ratios before and after dexamethasone administration. Repeated-measures ANCOVA revealed no significant main effect of day (F=0.00, df=1, 34, n.s.) or of group (F=0.44, df=1, 34, n.s.) and no significant group-by-day (F=0.04, df=1, 34, n.s.) or gender-by-day (F=0.22, df=1, 34, n.s.) interaction. However, the between-subjects analysis showed gender to be a significant covariate (F=13.90, df=1, 34, p=0.001) because of the lower predexamethasone ACTH-to-cortisol ratio in women (mean=1.05, SD=1.03; N=9) than in men (mean=2.87, SD=1.88; N=29). Dexamethasone level was not a significant covariate.
To further explore this gender effect, we performed a two-way ANOVA and ANCOVA, with both PTSD and gender as main effects, on the pre- and postdexamethasone ACTH-to-cortisol ratios, respectively. Results for both the basal ACTH-to-cortisol ratio (F=16.42, df=1, 34, p<0.0005) and the postdexamethasone ACTH-to-cortisol ratio (F=8.31, df=1, 33, p=0.007) showed significant main effects for gender, but neither demonstrated a significant group effect, and dexamethasone level was not a significant covariate in the postdexamethasone analysis. Finally, we examined the possibility of a gender-by-group interaction in a two-way repeated-measures analysis of pre- and postdexamethasone ACTH-to-cortisol ratios, which demonstrated only a between-subjects main effect of gender (F=13.34, df=1, 33, p=0.001) (i.e., for the average of the pre-and postdexamethasone ratios) without evidence of a significant PTSD effect, PTSD-by-gender interaction, or association with dexamethasone level as a covariate. There was no significant between-subjects effect of day or interaction with day.
Correlational Analyses
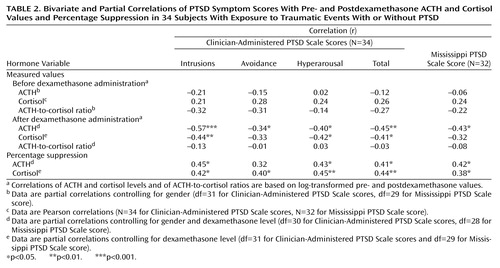
Correlations were used to examine associations among log-transformed biological measures, between these biological variables and the severity of PTSD symptoms, and between percentage suppression of ACTH or cortisol and symptom severity. There were significant associations between pre- and postdexamethasone cortisol levels (r=0.49, df=35, p=0.002), with dexamethasone concentration controlled for, and between pre- and postdexamethasone ACTH levels (r=0.59, df=34, p<0.0005), with gender and dexamethasone level controlled for. Additionally, postdexamethasone ACTH and cortisol levels were significantly correlated (r=0.42, df=34, p=0.02), with gender and dexamethasone level controlled for. In contrast, the predexamethasone ACTH and cortisol levels were not significantly correlated (r=0.10, df=35, n.s.), with gender effects controlled for. Percentage of ACTH suppression was significantly correlated with percentage of cortisol suppression (r=0.42, df=34, p=0.01), with gender and dexamethasone level controlled for.
Table 2 shows correlations between the biological variables and severity of PTSD symptoms for the 34 subjects who had been exposed to at least one potentially traumatic event and were interviewed with the Clinician-Administered PTSD Scale. Postdexamethasone ACTH and cortisol levels, but not predexamethasone values, were significantly correlated with PTSD symptoms. Both percentage of ACTH suppression and percentage of cortisol suppression were significantly correlated with severity of PTSD as represented by scores on the Mississippi PTSD Scale and the Clinician-Administered PTSD Scale. Neither the pre- nor postdexamethasone ACTH-to-cortisol ratio was significantly correlated with symptom severity (data not shown).
Discussion
The main findings of this study are that PTSD was associated with an enhanced suppression of ACTH in response to dexamethasone and that the ACTH-to-cortisol ratio did not differ between groups before or after dexamethasone administration. These findings suggest that it is unlikely that the low cortisol levels following dexamethasone administration in PTSD occur as a consequence of low adrenal output and/or abnormal adrenal sensitivity to ACTH. The data further strengthen the hypothesis that PTSD is associated with an enhanced cortisol negative feedback inhibition of ACTH secretion, likely mediated at the level of the pituitary, and implicate high pituitary glucocorticoid receptor sensitivity as a possible mechanism.
Enhanced cortisol suppression following dexamethasone administration has been consistently observed in PTSD (1, 13, 19–22). However, to our knowledge the ACTH response to dexamethasone in PTSD has not been previously investigated. There are several advantages to including measures of ACTH in studies using the DST. The ACTH response to dexamethasone is a more direct measure of feedback inhibition of the pituitary and, unlike cortisol, permits an estimation of feedback inhibition that is independent of the functioning of the adrenal gland. ACTH levels are also unaffected by artifacts associated with differences in corticosteroid binding globulin, which may affect the ratio of bound to unbound cortisol. High levels of corticosteroid binding globulin have been observed in PTSD (11) and might partially account for lower cortisol levels.
ACTH levels were measured at a single time point in the current study, which may be a methodological limitation because of the pulsatile secretion of this hormone and because of the moment-to-moment fluctuations that can occur in response to transient stressors, including the stress of venipuncture. However, the ACTH findings in the PTSD literature to date are based on just such samples. This study and the clear majority of studies have shown no significant difference in basal ACTH levels between PTSD and comparison subjects, even when cortisol levels obtained from the same blood sample were found to be significantly lower (11, 23, 24). Lower baseline 8:00 a.m. cortisol levels were also not observed in this study; however, this is consistent with results in other studies failing to demonstrate low cortisol levels in PTSD patients at 8:00 a.m. (e.g., reference 25) and, more important, with results from a comprehensive circadian rhythm analysis demonstrating that cortisol levels were significantly lower in PTSD patients than in comparison subjects at night, during the nadir of the diurnal cycle (8).
Although lower cortisol levels in the face of apparently normal ACTH levels could reflect a relatively low adrenal output, the lack of greater ACTH release in the setting of low cortisol levels implies that there may be an additional component of feedback to the pituitary acting to depress ACTH levels. High ACTH levels would be expected not only from the loss of feedback inhibition associated with low adrenal cortisol output but also from the possible contribution of greater CRH stimulation (5, 26). Thus, the consistent finding of apparently normal ACTH levels in PTSD suggests a more complex model of the regulatory influences on the pituitary in this disorder than is implied by low adrenal functioning, which may be in part accounted for by enhanced sensitivity to negative feedback.
In considering enhanced glucocorticoid negative feedback as a putative mechanism for the other HPA axis abnormalities observed in PTSD, it should be stated that glucocorticoid negative feedback inhibition is multifaceted, involving diverse levels of glucocorticoid action at multiple sites (e.g., pituitary, hypothalamus, and extrahypothalamic sites) and multiple receptor subtypes. Corticosteroids act on both mineralocorticoids and glucocorticoid receptors, which appear to mediate different components of feedback inhibition, and may produce different effects depending on factors such as time of day, in that receptor availability depends on whether glucocorticoid levels are at their peak or nadir (for example, see reference 3). The complexity in the regulation and action of corticosteroid receptors may result in some of the differential effects observed when somewhat different types of neuroendocrine challenge tests are used to test HPA axis functioning.
Indeed, neuroendocrine challenges involving the assessment of ACTH have produced mixed results in PTSD. In response to cholecystokinin tetrapeptide (CCK-4), a potent stimulator of ACTH, ACTH increases were less in individuals with PTSD than in comparison subjects, despite comparable ACTH levels at baseline (23). Cortisol levels were lower in the PTSD patients at baseline but rose to as high as those in the comparison subjects in response to CCK-4. It is important, however, that the rate of cortisol decline from the peak was faster in the subjects with PTSD, consistent with a more sensitive negative feedback inhibition that could be accounted for by greater glucocorticoid receptor responsiveness at the pituitary. In a second study (7), a high dose (2.5 g) of metyrapone was used to almost completely block cortisol production in both PTSD and comparison groups, thus removing negative feedback inhibition by cortisol from the HPA axis. Greater increases in ACTH and 11-deoxycortisol were observed in male combat veterans with PTSD than in nonexposed male subjects, suggesting that under resting conditions there is greater cortisol-induced negative feedback inhibition in PTSD subjects. Neither a pituitary nor adrenal insufficiency would likely result in a greater ACTH response to removal of negative feedback inhibition; the former would be associated with an attenuated ACTH response, and low adrenal output would not necessarily affect the ACTH response.
In contrast to the preceding findings, support for low adrenal functioning has also been observed. Kanter et al. (11) used lower doses of metyrapone (750 mg administered twice) to block endogenous cortisol production and then introduced cortisol by means of an infusion, to test the effects of negative feedback inhibition. The reintroduction of cortisol following metyrapone administration should result in a greater suppression of ACTH in PTSD if enhanced negative feedback were present. However, the ACTH response was not significantly different in individuals with and without PTSD. Interpretation of these findings is complicated, however, since there were group differences in the cortisol response to metyrapone. Metyrapone administration produced a significantly greater decrease in cortisol in the comparison subjects than the PTSD subjects. Thus, the higher endogenous cortisol levels may have inhibited ACTH secretion in the PTSD group.
The CRF challenge test has also produced discrepant results in PTSD. A blunted ACTH response to CRF, which is theoretically consistent with an enhanced negative feedback effect of cortisol to inhibit ACTH secretion, has been observed in combat veterans and in abused children. However, in sexually abused women with PTSD, an augmented ACTH response has been observed. There is some disagreement about whether the greater ACTH response to CRF occurs in the face of a low cortisol response to ACTH (27) or in tandem with an augmented cortisol response (28). The former pattern would be consistent with low adrenal functioning, but the latter would not.
Finally, it should be noted that a gender difference was observed with respect to basal and postdexamethasone levels of ACTH but not cortisol. Correspondingly, there was a significantly lower ratio of ACTH to cortisol in the women than in the men, consistent with the literature that suggests that the adrenal gland may be more sensitive to ACTH in women (29, 30). However, no significant gender differences were present in the percentage suppression of either cortisol or ACTH following dexamethasone administration. This observation requires further examination in a cohort with a sufficient proportion of women that the possibility of gender differences in the interaction between PTSD and the ACTH response to dexamethasone can be examined. It may be that yet-unidentified gender differences in ACTH release underlie some of the inconsistent neuroendocrine observations in PTSD.
In sum, the results of the current study suggest that the findings of enhanced suppression of cortisol in response to dexamethasone in PTSD may be mediated by dexamethasone effects on glucocorticoid receptors at the pituitary and are not a secondary consequence of a low adrenal capacity. In so doing, the data provide further support for the model of an enhanced negative feedback inhibition in PTSD. However, the findings should be considered in the light of a prior literature that has produced sometimes mixed support for this proposal. Although some of the discrepant findings may be explained by methodological problems or population differences, other neuroendocrine abnormalities, such as low pituitary or adrenal functioning or abnormal level of corticosteroid binding globulin, may also be part of the PTSD syndrome.
 |
 |
Received Feb. 11, 2003; revisions received May 6 and Oct. 7, 2003; accepted Oct. 31, 2003. From the Traumatic Stress Studies Program, Department of Psychiatry, Mount Sinai School of Medicine, and Bronx Veterans Affairs Medical Center, New York; the School of Psychology, University of Reading, Reading, U.K.; and the Department of Neuroscience, Douglas Hospital Research Centre, Montreal. Address reprint requests to Dr. Yehuda, Bronx VA OOMH, 130 West Kingsbridge Rd., Bronx, NY 10468; [email protected] (e-mail). Supported by NIMH grant R01 MH-49555 (Dr. Yehuda) and a VA Merit Review grant (Dr. Yehuda).

Figure 1. Plasma ACTH Levels at 8:00 a.m. on Two Days, Before and After Ingestion of 0.5 mg of Dexamethasone, in 38 Subjects With and Without PTSDa
aBlack lines represent mean values.
1. Yehuda R, Boisoneau D, Lowy MT, Giller EL: Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry 1995; 52:583–593Crossref, Medline, Google Scholar
2. Ribeiro SC, Tandon R, Grunhaus L, Greden JF: The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry 1993; 150:1618–1629Link, Google Scholar
3. de Kloet ER, Joels M, Oitzl M, Sutanto W: Implication of brain corticosteroid receptor diversity for the adaptation syndrome concept. Methods Achiev Exp Pathol 1991; 14:104–132Medline, Google Scholar
4. Cole MA, Kim PJ, Kalman BA, Spencer RL: Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology 2000; 25:151–167Crossref, Medline, Google Scholar
5. Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS: Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997; 154:624–629Link, Google Scholar
6. Smith MA, Davidson J, Ritchie JC, Kudler H, Lipper S, Chappell P, Nemeroff CB: The corticotropin-releasing hormone test in patients with posttraumatic stress disorder. Biol Psychiatry 1989; 26:349–355Crossref, Medline, Google Scholar
7. Yehuda R, Levengood RA, Schmeidler J, Wilson S, Guo LS, Gerber D: Increased pituitary activation following metyrapone administration in post-traumatic stress disorder. Psychoneuroendocrinology 1996; 21:1–16Crossref, Medline, Google Scholar
8. Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ: Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatry 1996; 40:79–88Crossref, Medline, Google Scholar
9. Rasmusson AM, Lipschitz DS, Wang S, Hu S, Vojvoda D, Bremner JD, Southwick SM, Charney DS: Increased pituitary and adrenal reactivity in premenopausal women with posttraumatic stress disorder. Biol Psychiatry 2001; 50:965–977Crossref, Medline, Google Scholar
10. Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, Miller AH, Nemeroff CB: Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 2000; 284:592–597Crossref, Medline, Google Scholar
11. Kanter ED, Wilkinson CW, Radant AD, Petrie EC, Dobie DJ, McFall ME, Peskind ER, Raskind MA: Glucocorticoid feedback sensitivity and adrenocortical responsiveness in posttraumatic stress disorder. Biol Psychiatry 2001; 50:238–245Crossref, Medline, Google Scholar
12. Yehuda R: Current status of cortisol findings in post-traumatic stress disorder. Psychiatr Clin North Am 2002; 25:341–368, viiCrossref, Medline, Google Scholar
13. Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C: The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biol Psychiatry 2002; 52:393–403Crossref, Medline, Google Scholar
14. Green BL: Trauma History Questionnaire, in Measurement of Stress, Trauma, and Adaptation. Edited by Stamm BH. Lutherville, Md, Sidran, 1996, pp 366–369Google Scholar
15. Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, Keane TM: The development of a Clinician-Administered PTSD Scale. J Trauma Stress 1995; 8:75–90Crossref, Medline, Google Scholar
16. First MB, Spitzer RL, Gibbon M, Williams JBW: Structured Clinical Interview for DSM-IV Axis I Disorders (SCID). New York, New York State Psychiatric Institute, Biometrics Research, 1995Google Scholar
17. Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL, Mason JW: Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis 1990; 178:366–369Crossref, Medline, Google Scholar
18. Lowy MT, Meltzer HY: Dexamethasone bioavailability: implications for DST research. Biol Psychiatry 1987; 22:373–385Crossref, Medline, Google Scholar
19. Yehuda R, Southwick SM, Krystal JH, Bremner D, Charney DS, Mason JW: Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am J Psychiatry 1993; 150:83–86Link, Google Scholar
20. Kellner M, Baker DG, Yehuda R: Salivary cortisol in Operation Desert Storm returnees. Biol Psychiatry 1997; 42:849–850Crossref, Medline, Google Scholar
21. Goenjian AK, Yehuda R, Pynoos RS, Steinberg AM, Tashjian M, Yang RK, Najarian LM, Fairbanks LA: Basal cortisol, dexamethasone suppression of cortisol, and MHPG in adolescents after the 1988 earthquake in Armenia. Am J Psychiatry 1996; 153:929–934Link, Google Scholar
22. Stein MB, Yehuda R, Koverola C, Hanna C: Enhanced dexamethasone suppression of plasma cortisol in adult women traumatized by childhood sexual abuse. Biol Psychiatry 1997; 42:680–686Crossref, Medline, Google Scholar
23. Kellner M, Wiedemann K, Yassouridis A, Levengood R, Guo LS, Holsboer F, Yehuda R: Behavioral and endocrine response to cholecystokinin tetrapeptide in patients with posttraumatic stress disorder. Biol Psychiatry 2000; 47:107–111Crossref, Medline, Google Scholar
24. Hockings GI, Grice JE, Ward WK, Walters MM, Jensen GR, Jackson RV: Hypersensitivity of the hypothalamic-pituitary-adrenal axis to naloxone in post-traumatic stress disorder. Biol Psychiatry 1993; 33:585–593Crossref, Medline, Google Scholar
25. Thaller V, Vrkljan M, Hotujac L, Thakore J: The potential role of hypocortisolism in the pathophysiology of PTSD and psoriasis. Coll Antropol 1999; 23:611–619Medline, Google Scholar
26. Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD Jr: Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999; 156:585–588; correction, 156:986Abstract, Google Scholar
27. Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB: Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry 2001; 158:575–581Link, Google Scholar
28. Rasmusson AM, Lipschitz DS, Wang S, Hu S, Vojvoda D, Bremner JD, Southwick SM, Charney DS: Increased pituitary and adrenal reactivity in premenopausal women with posttraumatic stress disorder. Biol Psychiatry 2001; 50:965–977Crossref, Medline, Google Scholar
29. Roelfsema F, van den Berg G, Frolich M, Veldhuis JD, van Eijk A, Buurman MM, Etman BH: Sex-dependent alteration in cortisol response to endogenous adrenocorticotropin. J Clin Endocrinol Metab 1993; 77:234–240Medline, Google Scholar
30. Horrocks PM, Jones AF, Ratcliffe WA, Holder G, White A, Holder R, Ratcliffe JG, London DR: Patterns of ACTH and cortisol pulsatility over twenty-four hours in normal males and females. Clin Endocrinol (Oxf) 1990; 32:127–134Crossref, Medline, Google Scholar

