
Amyloid β Pathology in Alzheimer’s Disease and Schizophrenia
Abstract
OBJECTIVE: Severe cognitive impairment is common in elderly patients with schizophrenia. Alzheimer’s disease is the main cause of dementia among the elderly. Biochemical and genetic studies suggest that amyloid β-peptide is central in Alzheimer’s disease. The authors examined the possible involvement of amyloid β-peptide in cognitive impairment in schizophrenia. METHOD: Specific antibodies against two major forms of amyloid β-peptide, Aβx-40 and Aβx-42, were used in sandwich enzyme-linked immunosorbent assays to determine the levels of amyloid β-peptide in postmortem brain samples from Alzheimer’s disease patients (N=10), normal elderly comparison subjects (N=11), and schizophrenia patients with (N=7) or without (N=26) Alzheimer’s disease. RESULTS: The levels of amyloid β-peptide were highest in the Alzheimer’s disease patients, followed by the patients with schizophrenia and comparison subjects. The mean Aβx-42 level in the schizophrenia patients without Alzheimer’s disease was similar to that in the comparison subjects, but the level in the schizophrenia patients with Alzheimer’s disease was significantly higher than in those without Alzheimer’s disease or the comparison subjects. The Aβx-42 level in the schizophrenia patients with Alzheimer’s disease was significantly lower than the level in the Alzheimer’s disease cohort. CONCLUSIONS: In contrast to elderly schizophrenia patients with Alzheimer’s disease pathology, those without Alzheimer’s disease had amyloid β-peptide levels that were not significantly different from those of normal subjects; hence amyloid β-peptide does not account for the cognitive deficits in this group. These results suggest that the causes of cognitive impairment in “pure” schizophrenia are different from those in Alzheimer’s disease.
Clinical studies (1–5) suggest that severe cognitive impairment is common among institutionalized elderly patients with schizophrenia. The cognitive impairment seen in these patients is progressive and is not attributable to a lack of cooperation, attention, or motivation or to neuroleptic treatments (6). Since Alzheimer’s disease is the most common cause of dementia in the elderly, it is a plausible candidate as a comorbid disease contributing to the dementia observed in elderly patients with schizophrenia. Postmortem neuropathologic investigations (7–10) have provided conflicting results about the occurrence of Alzheimer’s disease in elderly patients with schizophrenia. In the most recent studies (1, 11, 12) no notable neuropathologic abnormalities were seen that could be implicated as the underlying cause of cognitive impairment.
Alzheimer’s disease patients often have psychiatric manifestations of disease, such as psychosis (e.g., delusions and hallucinations) and disruptive behaviors (e.g., psychomotor agitation and physical aggression), especially in the later stages of the disease (13–15). It has been shown (16–19) that psychosis and aggression are associated with more rapid rates of cognitive and functional progression of Alzheimer’s disease.
Thus, although schizophrenia and Alzheimer’s disease may not share common neuropathologic lesions, such as neuritic plaques, the commonality of some cognitive and psychiatric symptoms in the two diseases suggests the potential existence of overlapping molecular pathogenic pathways in Alzheimer’s disease and schizophrenia. Alzheimer’s disease is a progressive, neurodegenerative disease characterized by loss of functioning and death of neurons in several areas of the brain (20). Deposition of the amyloid β-peptide into plaques in the brain parenchyma and cerebral blood vessel walls, as well as accumulation of neurofibrillary tangles in neurons, neuronal loss, and synaptic pathology, are all distinguishing features of Alzheimer’s disease. The senile plaques consist of an amyloid core, which is stained by the β-sheet-specific dye Congo red. The main protein component of the plaques is Aβ0. There are two major forms of amyloid β-peptide, the 40-residue Aβx-40 and the 42-residue Aβx-42; the latter is more amyloidogenic (21). These peptides are proteolytic fragments of the Alzheimer amyloid precursor protein, a class I transmembrane glycoprotein expressed throughout most tissues in the body. Genetic and biochemical evidence implicates a central role for amyloid β-peptide in the pathophysiology of Alzheimer’s disease (22), and evidence has shown that the density of plaques (23) and the levels of amyloid β-peptide (24) correlate with the severity of dementia. Since even amyloid β-peptide deposits that are not associated with classical neuritic plaques (diffuse plaques) can correlate significantly with mild dementia in the elderly (25), it is possible that elevations in the levels of the nonaggregated form(s) of amyloid β-peptide can contribute to cognitive deficits in schizophrenia patients in the absence of the Alzheimer’s-associated neuritic plaques with amyloid cores.
We investigated whether cognitive decline in elderly patients with schizophrenia is related to amyloid β-peptide by measuring the levels of total amyloid, Aβx-40, and Aβx-42 in the dorsolateral prefrontal cortex of patients with schizophrenia.
Method
Human Postmortem Tissue
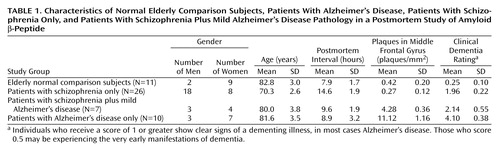
We categorized frozen postmortem brain samples into four groups: normal elderly comparison subjects (N=11), patients with Alzheimer’s disease (N=10), patients with schizophrenia only (N=26), and patients with schizophrenia and mild Alzheimer’s disease pathology (N=7). The mean age, postmortem interval, and sex distribution of each cohort are shown in Table 1. The brain specimens were obtained from the Mount Sinai School of Medicine Department of Psychiatry Brain Bank. Each brain was divided midsagittally at autopsy. The right half was fixed in 4% paraformaldehyde for neuropathologic assessment, and the left half was dissected into approximately 8-mm coronal sections, flash frozen in liquid nitrogen, and stored at –80°C until dissection. For this study, the dorsolateral prefrontal cortex (Brodmann’s area 46) (1, 26–28) was dissected from the frozen sections without thawing and was pulverized into the consistency of fine powder and divided into aliquots. We used 25-mg aliquots for the assays described.
Each selected brain was neuropathologically assessed, and the medical and psychiatric histories were reviewed in detail (1). All assessment and postmortem evaluations and procedures were approved by the institutional review boards of Pilgrim Psychiatric Center, Mount Sinai School of Medicine and the Bronx VA Medical Center. Written informed consent was obtained from the relatives of the study subjects after the procedures had been fully explained. Antemortem assessment and postmortem chart reviews were used to determine the degree of dementia present in each case (1, 23, 24) according to the Clinical Dementia Rating (5, 29).
The normal elderly comparison subjects showed no history of neuropsychiatric disease, were not demented (Clinical Dementia Rating, ≤0.5), and were free of any discernible neuropathologic lesions. All of the schizophrenia subjects had been chronically hospitalized at Pilgrim Psychiatric Center (New York City) and met the DSM-III-R criteria for schizophrenia according to antemortem assessment and postmortem chart review (1, 5, 30, 31). The patients with Alzheimer’s disease had been residents of the Jewish Home and Hospital in New York City (an affiliate of the Mount Sinai School of Medicine) and met the criteria for definite Alzheimer’s disease of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) (32) and Khachaturian (33). Subjects classified as having schizophrenia with mild Alzheimer’s disease neuropathology met the preceding criteria for schizophrenia but also evidenced Alzheimer’s-related lesions of sufficient magnitude to receive a neuropathologic diagnosis of possible Alzheimer’s disease (according to the CERAD criteria). In addition to the semiquantitative ratings of neuritic plaques required by the CERAD protocol, the numbers of neuritic plaques in representative sections of five neocortical areas, including the middle frontal gyrus, were counted in each case as described previously (23). Estimates of neuritic plaque density in the middle frontal gyrus (the area corresponding to the region dissected for measurement of amyloid β-peptide immunoreactivity) were then used in the selection of subjects as follows: normal comparison and schizophrenia cases were selected to evidence no more than 2 neuritic plaques per square millimeter, cases of schizophrenia with mild Alzheimer’s disease pathology were selected to have neuritic plaque densities of more than 2 but fewer than 8 plaques per square millimeter, and Alzheimer’s disease cases were selected to have 8 or more neuritic plaques per square millimeter.
Amyloid β-Peptide Extraction and Sample Preparation
The frozen powdered tissue homogenates (25 mg) from the dorsolateral prefrontal cortex were homogenized by sonication in 800 μl of phosphate-buffered saline solution containing protease inhibitor cocktail. The homogenates were centrifuged at 100,000 g for 60 minutes at 4°C. The pellets were delipidated by a modification of an established protocol for chloroform-methanol extraction (34). Briefly, homogenization of the pellets in 400 μl of methanol was followed by addition of 300 μl of chloroform and 500 μl of water. The samples were centrifuged at 10,000 g for 2 minutes, and the aqueous phase was removed. The protein-containing interphase was pelleted, after addition of another 400 μl of methanol, by centrifugation at 10,000 g for 5 minutes. Amyloid β-peptide was extracted from the pellets by sonication and mixing by use of vortex (10–15 minutes) in 150 μl of 70% formic acid containing 100 mM of betaine. The formic acid fractions containing amyloid β-peptide were collected from the clear supernatant resulting from centrifugation of the extract at 100,000 g for 30 minutes at 4°C and saved for analyses. Aliquots (30 μl) of each formic acid extract were neutralized in 510 μl of 1 M Tris HCl and 100 mM of betaine, pH 10.3, and particulate matter was suspended by sonication. The samples were further diluted 1:50 in enzyme-linked immunosorbent assay (ELISA) capture and wash buffer (phosphate-buffered saline solution, 5 mM of EDTA, 2 mM of betaine, 0.1% bovine serum albumin, 0.05% Tween–20, 0.2% CHAPS [3-([3-cholamidopropyl] dimethylammonio)-1-propanesulfonic acid], and 0.05% NaN3) before the ELISA measurements.
Sandwich ELISAs
Peptides containing N-terminal cysteine residues corresponding to the six C-terminal residues of Aβx-40 and Aβx-42 were synthesized, conjugated to keyhole limpet hemocyanin, and injected into rabbits. Serum obtained from the rabbits was analyzed for specificity and cross-reactivity by spot blotting, immunoblotting, and peptide ELISAs. After this primary screening, selected antisera were affinity-purified on resins to which the peptides used as antigens had been coupled. One antibody for Aβx-40 and one antibody for Aβx-42 were selected and used throughout the study.
Microtiter plates were coated at 4°C for 36 hours with 5 μg/ml of monoclonal antibody 6E10 diluted in phosphate-buffered saline solution. The antibody 6E10 recognizes an epitope between 5 and 10 residues of amyloid β-peptide, which allows the detection of amyloid β-peptide beginning at residue 1 but also of different N-terminally truncated amyloid β-peptide species. The coated plates were washed four times with phosphate-buffered saline solution containing 0.05% Tween-20. Unoccupied binding sites on the plates were blocked by incubating the plates with ELISA blocking reagent for 1 hour at room temperature. The plates were washed four times with phosphate-buffered saline solution containing 0.05% Tween-20, and standards and samples were applied to appropriate wells. Synthetic Aβ1-40 and Aβ1-42 were used as standard peptides and stored in aliquots at –80°C (100 μg/ml in hexafluoroisopropanol). Standards were applied in duplicates and samples in quadruplicates before incubation for 24 hours at 4°C. After the capture phase, the plates were washed four times with phosphate-buffered saline solution containing 0.05% Tween-20, and appropriate amyloid β-peptide end-specific antibodies were added. The plates were incubated overnight at 4°C and then washed four times with phosphate-buffered saline solution containing 0.05% Tween-20. Reporter antibody, horseradish-peroxidase-linked antirabbit IgG was added (0.25 μg/ml), and the plates were incubated for 1 hour at room temperature. The plates were washed four times in phosphate-buffered saline solution containing 0.05% Tween-20 and developed by using o-phenylenediamine as substrate. The o-phenylenediamine tablets were dissolved in 0.05 M phosphate-citrate buffer, and fresh 30% hydrogen peroxide was added. Reactions were stopped by using 2 M sulfuric acid after sufficient color development. The plates were analyzed in a 96-well reader at 490 nm. The detection limit for synthetic Aβx-40 and Aβx-42 was 1 pM/liter. All samples were analyzed in the linear range of the ELISA assay.
Total amyloid β-peptide ELISA was performed essentially as described in the preceding. Aβ1-40 was used as standard peptide, monoclonal antibody 4G8 was used as secondary antibody, and horseradish-peroxidase-conjugated avidin was used as reporter antibody, the latter two at dilutions of 1:2000 and 1:5000, respectively.
Statistical Analysis
Analysis of variance (ANOVA) followed by Newman-Keuls tests were used to analyze the results of these studies. ANOVA was used for data from the entire group. Post hoc analyses were used to compare differences between groups. Statistical analyses were performed with Statistica (release 5.5, Statsoft, Tulsa, Okla.).
Results
Demographic characteristics of the cohort are shown in Table 1. The four groups did not differ significantly from each other with respect to age at death (F=2.7, df=3, 53, p=0.06) or postmortem interval (F=2.7, df=3, 53, p=0.06). Individual ANOVAs performed for total amyloid β-peptide, Aβx-40, and Aβx-42 levels revealed significant group differences (Figure 1). Total amyloid β-peptide immunoreactivity was significantly higher in the dorsolateral prefrontal cortex of the Alzheimer’s disease group than in the other groups overall (F=17.5, df=3, 50, p<0.0001) and in each group individually (Newman-Keuls tests, p<0.0009). The patients with schizophrenia, with or without Alzheimer’s-like neuropathology, did not differ significantly from the comparison subjects with respect to total amyloid β-peptide immunoreactivity (Newman-Keuls tests, p>0.2). Nearly identical results were obtained from the analysis of Aβx-40 immunoreactivity (F=17.5, df=3, 50, p<0.00001; Newman-Keuls tests, p<0.0003). A different pattern of results emerged from examination of Aβx-42 immunoreactivity. ANOVA revealed a significant effect of group (F=29.2, df=3, 50, p<0.00001). As expected, post hoc tests (Newman-Keuls) showed that the levels of Aβx-42 were significantly higher in the dorsolateral prefrontal cortex of the Alzheimer’s disease group than in each of the other groups (p<0.0002). In contrast to the pattern of results for total amyloid β-peptide and Aβx-40 immunoreactivity, the levels of Aβx-42 were significantly higher in the dorsolateral prefrontal cortex of the schizophrenia patients with comorbid possible Alzheimer’s disease than in the normal comparison subjects (Newman-Keuls test, p=0.003) and the schizophrenia patients with no neuropathology (Newman-Keuls test, p=0.006). The levels of Aβx-42 in this group were intermediate between those of the normal comparison subjects and the patients with Alzheimer’s disease only. The results for direct counts of neuritic plaque density (Table 1) were similar to the results for Aβx-42 (F=135.6, df=3, 50, p<0.0001). The Alzheimer’s disease group differed significantly from each of the other groups (Newman-Keuls tests, p<0.0001). Levels of total amyloid β-peptide, Aβx-40, and Aβx-42 were also significantly correlated with Clinical Dementia Rating scores when the entire cohort was included in the analysis (r=0.38, r=0.43, and r=0.50, respectively, N=54, p<0.004) and with measures of neuritic plaque density (r=0.53, r=0.54, r=0.77, respectively, N=54, p<0.0001).
Discussion
The role of amyloid β-peptide in the pathogenesis of neurodegenerative disorders is not completely elucidated, but its toxic effect is not necessarily correlated with senile plaque deposition, since it has been shown that the neurotoxic effect of amyloid β-peptide is independent of plaque formation in transgenic mouse models (35). It has been suggested that neurotoxic effects can be induced by diffusible amyloid β-peptide oligomers (36) or by intraneuronal accumulation of amyloid β-peptide (37). Regardless of the finer molecular details and oligomeric assembly of neurotoxic amyloid β-peptide species, we recently showed that Aβx-40 and Aβx-42 correlate with the severity of dementia in Alzheimer’s disease (24). Hence, biochemically determined levels of brain amyloid β-peptide are good predictors of dementia severity in Alzheimer’s disease irrespective of their role in neurodegeneration and their intra–/extracellular location. This study provides evidence that elderly patients with schizophrenia showing Alzheimer’s disease neuropathology in the brain have increased levels of Aβ0. Thus, cognitive impairment in these patients could be related to the dementia-associated amyloid β-peptide pathogenicity in the brain. However, the larger group of elderly patients with schizophrenia, who were comparably demented but did not evidence Alzheimer’s-related histopathology, did not show significantly elevated levels of amyloid β-peptide. Analysis of the total brain amyloid β-peptide content in the present patient cohort showed that the occurrence of cognitive decline in the course of schizophrenia is distinct from the neuropathologic or molecular processes linked to amyloid β-peptide in Alzheimer’s disease.
The results presented here can, at least partially, address the hypothesis that some neuroleptics used for schizophrenia protect against the development of Alzheimer’s disease (38). It has been demonstrated (39) that haloperidol, frequently used as antipsychotic medication, and the related compound droperidol inhibit the production of amyloid β-peptide by cultured human neuronal and nonneuronal cells. The inhibition was concentration dependent, and the lowest active medication dose tested approached physiological concentrations of amyloid β-peptide. The inhibitory effect of these compounds on amyloid β-peptide processing was consistent with the previously identified proteolytic inhibitory activity of haloperidol (40). Arguing against this hypothesis, however, was the failure of any of the measures of amyloid β-peptide to correlate significantly (r=0.003 and r=–0.03 for Aβx-40 and Aβx-42, respectively) with the number of weeks that the schizophrenia subjects had been free from neuroleptic medications before their deaths (range=0–125 weeks). Given the likelihood that amyloid β-peptide accumulation and plaque formation occur over a prolonged period, 125 weeks without neuroleptics may not have been long enough to overcome the inhibitory consequences of years of exposure to therapeutic doses of antipsychotic medications.
There are also several studies supporting an influence of nicotine on the processing of amyloid precursor proteins and on production of amyloid β-peptide. Patients with schizophrenia are commonly heavy smokers (prevalence between 74% and 92%) (41), and nicotine can decrease amyloid β-peptide toxicity and fibril formation (42). It has been proposed that nicotine or nicotinic receptor agonists might improve cognitive functioning not only by supplementing cholinergic neurotransmission but also by protecting against amyloid β-peptide neurotoxicity, probably through increased release of amyloid precursor proteins after activation of nicotinic receptors (43). Reliable data on smoking history were not available for the entire study cohort, and so we cannot definitively analyze nicotinic influences in this study. However, review of medical records enabled us to identify seven schizophrenia patients whose medical records documented that they were nonsmokers and nine schizophrenia patients with documented evidence of tobacco smoking until shortly before death. Analysis of total amyloid β-peptide, Aβx-40, and Aβx-42 did not reveal statistically significant differences between the nonsmokers and smokers, but the mean total amyloid β-peptide level was nominally higher in the cortexes of nonsmokers (mean=1464.5 pmol/g tissue, SD=126) than in the smokers (mean=442.7, SD=136) (t=1.9, df=14, p=0.07).
Currently, experimental therapy for Alzheimer’s disease is aimed at decreasing amyloid β-peptide levels in the brain to preserve cognitive functioning. According to our findings, although schizophrenia patients with comorbid Alzheimer’s disease neuropathology are likely to benefit from emerging therapies based on amyloid β-peptide, most other elderly schizophrenia patients with dementia are unlikely to benefit from such therapies.
In summary, there is still a need for a biological model explaining the underlying pathogenesis of the cognitive impairment in elderly patients with schizophrenia. Our study suggests that amyloid β-peptide is not a causative agent for cognitive decline in this particular patient group. However, the cause-and-effect relationship between amyloid β-peptide formation, processing of amyloid precursor proteins, and brain dysfunction in schizophrenia needs further studies.
 |
Received June 5, 2002; revision received Nov. 13, 2002; accepted Nov. 25, 2002. From the Department of Clinical Neuroscience, Occupational Therapy and Elderly Care Research, Karolinska Institutet; the Department of Neurodegenerative Disorders, Medical Research Centre, Polish Academy of Sciences, Warsaw; and the Department of Psychiatry, Mount Sinai School of Medicine and Bronx Veterans Affairs Medical Center, Bronx, N.Y. Address reprint requests to Dr. Religa, Department of Clinical Neuroscience, Occupational Therapy and Elderly Care Research, Karolinska Institutet, 141 86 Huddinge, Sweden; [email protected] (e-mail). Supported by the Swedish Institute (New Visby Programme), by Sweden’s Royal Academy of Sciences, by the Polish Academy of Sciences (Drs. Religa and Styczynska), and by an award from the National Alliance for Research on Schizophrenia and Depression (Dr. Haroutunian).

Figure 1. Mean Postmortem Levels of Total Amyloid β-Peptide, Aβx-40, and Aβx-42 in the Dorsolateral Prefrontal Cortex of Normal Elderly Comparison Subjects, Patients With Alzheimer’s Disease, Patients With Schizophrenia Only, and Patients With Schizophrenia Plus Mild Alzheimer’s Disease Pathology
aSignificantly different from all other groups (Newman-Keuls tests, p<0.01).
bSignificantly different from all other groups (Newman-Keuls tests, p<0.001).
1. Purohit DP, Perl DP, Haroutunian V, Powchik P, Davidson M, Davis KL: Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry 1998; 55:205-211Crossref, Medline, Google Scholar
2. Buhrich N, Crow TJ, Johnstone EC, Owens DG: Age disorientation in chronic schizophrenia is not associated with pre-morbid intellectual impairment or past physical treatment. Br J Psychiatry 1988; 152:466-469Crossref, Medline, Google Scholar
3. Johnstone EC, Owens DG, Gold A, Crow TJ, MacMillan JF: Institutionalization and the defects of schizophrenia. Br J Psychiatry 1981; 139:195-203Crossref, Medline, Google Scholar
4. Taylor MA, Abrams R: Cognitive impairment in schizophrenia. Am J Psychiatry 1984; 141:196-201Link, Google Scholar
5. Davidson M, Harvey PD, Powchik P, Parrella M, White L, Knobler HY, Losonczy MF, Keefe RSE, Katz S, Frecska E: Severity of symptoms in chronically institutionalized geriatric schizophrenic patients. Am J Psychiatry 1995; 152:197-207Link, Google Scholar
6. Harvey PD, Davidson M, Powchik P, Parrella M, White L, Mohs RC: Assessment of dementia in elderly schizophrenics with structured rating scales. Schizophr Res 1992; 7:85-90Crossref, Medline, Google Scholar
7. Arnold SE, Franz BR, Trojanowski JQ: Elderly patients with schizophrenia exhibit infrequent neurodegenerative lesions. Neurobiol Aging 1994; 15:299-303Crossref, Medline, Google Scholar
8. Buhl L, Bojsen-Moller M: Frequency of Alzheimer’s disease in a postmortem study of psychiatric patients. Dan Med Bull 1988; 35:288-290Medline, Google Scholar
9. Prohovnik I, Dwork AJ, Kaufman MA, Willson N: Alzheimer-type neuropathology in elderly schizophrenia patients. Schizophr Bull 1993; 19:805-816Crossref, Medline, Google Scholar
10. Purohit DP, Davidson M, Perl DP, Powchik P, Haroutunian VH, Bierer LM, McCrystal J, Losonczy M, Davis KL: Severe cognitive impairment in elderly schizophrenic patients: a clinicopathological study. Biol Psychiatry 1993; 33:255-260Crossref, Medline, Google Scholar
11. Powchik P, Davidson M, Haroutunian V, Gabriel SM, Purohit DP, Perl DP, Harvey PD, Davis KL: Postmortem studies in schizophrenia. Schizophr Bull 1998; 24:325-341Crossref, Medline, Google Scholar
12. Dwork AJ, Susser ES, Keilp J, Waniek C, Liu D, Kaufman M, Zemishlany Z, Prohovnik I: Senile degeneration and cognitive impairment in chronic schizophrenia. Am J Psychiatry 1998; 155:1536-1543Link, Google Scholar
13. Reisberg B, Borenstein J, Salob SP, Ferris SH, Franssen E, Georgotas A: Behavioral symptoms in Alzheimer’s disease: phenomenology and treatment. J Clin Psychiatry 1987; 48(May suppl):9-15Google Scholar
14. Rubin EH, Morris JC, Berg L: The progression of personality changes in senile dementia of the Alzheimer’s type. J Am Geriatr Soc 1987; 35:721-725Crossref, Medline, Google Scholar
15. Devanand DP, Jacobs DM, Tang MX, Del Castillo-Castaneda C, Sano M, Marder K, Bell K, Bylsma FW, Brandt J, Albert M, Stern Y: The course of psychopathologic features in mild to moderate Alzheimer disease. Arch Gen Psychiatry 1997; 54:257-263Crossref, Medline, Google Scholar
16. Stern Y, Tang MX, Albert MS, Brandt J, Jacobs DM, Bell K, Marder K, Sano M, Devanand D, Albert SM, Bylsma F, Tsai WY: Predicting time to nursing home care and death in individuals with Alzheimer disease. JAMA 1997; 277:806-812Crossref, Medline, Google Scholar
17. Mortimer JA, Ebbitt B, Jun SP, Finch MD: Predictors of cognitive and functional progression in patients with probable Alzheimer’s disease. Neurology 1992; 42:1689-1696Crossref, Medline, Google Scholar
18. Mega MS, Cummings JL, Fiorello T, Gornbein J: The spectrum of behavioral changes in Alzheimer’s disease. Neurology 1996; 46:130-135Crossref, Medline, Google Scholar
19. Jost BC, Grossberg GT: The evolution of psychiatric symptoms in Alzheimer’s disease: a natural history study. J Am Geriatr Soc 1996; 44:1078-1081Crossref, Medline, Google Scholar
20. Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999; 399(6738 suppl):A23-A31Crossref, Medline, Google Scholar
21. Jarrett JT, Lansbury PT Jr: Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993; 73:1055-1058Crossref, Medline, Google Scholar
22. Hardy J: Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 1997; 20:154-159Crossref, Medline, Google Scholar
23. Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC: Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol 1998; 55:1185-1191Crossref, Medline, Google Scholar
24. Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD: Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 2000; 283:1571-1577Crossref, Medline, Google Scholar
25. Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA, Berg L: Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996; 46:707-719Crossref, Medline, Google Scholar
26. Rajkowska G, Goldman-Rakic PS: Cytoarchitectonic definition of prefrontal areas in the normal human cortex, I: remapping of areas 9 and 46 using quantitative criteria. Cereb Cortex 1995; 5:307-322Crossref, Medline, Google Scholar
27. Rajkowska G, Goldman-Rakic PS: Cytoarchitectonic definition of prefrontal areas in the normal human cortex, II: variability in locations of areas 9 and 46 and relationship to the Talairach coordinate system. Cereb Cortex 1995; 5:323-337Crossref, Medline, Google Scholar
28. Dracheva S, Marras SAE, Elhakem SL, Kramer FR, Davis KL, Haroutunian V: N-Methyl-d-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am J Psychiatry 2001; 158:1400-1410; correction, 158:2107Link, Google Scholar
29. Morris JC: The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43:2412-2414Crossref, Medline, Google Scholar
30. Harvey PD, Howanitz E, Parrella M, White L, Davidson M, Mohs RC, Hoblyn J, Davis KL: Symptoms, cognitive functioning, and adaptive skills in geriatric patients with lifelong schizophrenia: a comparison across treatment sites. Am J Psychiatry 1998; 155:1080-1086Link, Google Scholar
31. Harvey PD, Parrella M, White L, Mohs RC, Davidson M, Davis KL: Convergence of cognitive and adaptive decline in late-life schizophrenia. Schizophr Res 1999; 35:77-84Crossref, Medline, Google Scholar
32. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L: The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41:479-486Crossref, Medline, Google Scholar
33. Khachaturian Z: Diagnosis of Alzheimer’s disease. Arch Neurol 1985; 42:1097-1105Crossref, Medline, Google Scholar
34. Wessel D, Flugge UI: A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 1984; 138:141-143Crossref, Medline, Google Scholar
35. Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 1999; 96:3228-3233Crossref, Medline, Google Scholar
36. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998; 95:6448-6453Crossref, Medline, Google Scholar
37. Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT Jr, Kosik KS: A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med 1998; 4:730-734Crossref, Medline, Google Scholar
38. Arnold SE, Trojanowski JQ, Gur RE, Blackwell P, Han LY, Choi C: Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry 1998; 55:225-232Crossref, Medline, Google Scholar
39. Higaki J, Murphy GM Jr, Cordell B: Inhibition of beta-amyloid formation by haloperidol: a possible mechanism for reduced frequency of Alzheimer’s disease pathology in schizophrenia. J Neurochem 1997; 68:333-336Crossref, Medline, Google Scholar
40. DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babe LM, Craik CS: Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc Natl Acad Sci USA 1990; 87:6644-6648Crossref, Medline, Google Scholar
41. Adler LE, Olincy A, Waldo M, Harris JG, Griffith J, Stevens K, Flach K, Nagamoto H, Bickford P, Leonard S, Freedman R: Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull 1998; 24:189-202Crossref, Medline, Google Scholar
42. Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG: Nicotine inhibits amyloid formation by the Aβ-peptide. Biochemistry 1996; 35:13568-13578Crossref, Medline, Google Scholar
43. Seo J, Kim S, Kim H, Park CH, Jeong S, Lee J, Choi SH, Chang K, Rah J, Koo J, Kim E, Suh Y: Effects of nicotine on APP secretion and Aβ- or CT(105)-induced toxicity. Biol Psychiatry 2001; 49:240-247Crossref, Medline, Google Scholar

