
Will the Genomics Revolution Revolutionize Psychiatry?
At the time DNA was discovered 50 years ago, psychiatric genetics was in a state of relative dormancy, resulting from the misuse of genetic theories during the World War II era. As the excitement generated by the “new genetics” spread across medical specialty areas, the second half of the century was declared the century of the biological sciences, and genetics research blossomed (1). Dismissing the widespread notion of a basic conflict between human genetics and religious tenets, Pope Pius XII gave a policy-setting address in 1953, encouraging the need for systematic and ideologically unshackled research in human genetics (1). In the same year as the discovery of DNA, Franz Kallman’s review of progress in psychiatric genetics, published in TheAmerican Journal of Psychiatry, described a sophisticated series of twin and family studies in the United States (1–3) and Europe (4, 5) that corroborated the genetic roots of schizophrenia and manic depressive psychosis that had been demonstrated in the early part of the 20th century. Subsequent research has continued to expand our knowledge of the genetics of psychiatric disorders. As the classification system has grown more specific, genetic investigations have continued to demonstrate the importance of familial and genetic factors underlying most of the major psychiatric conditions.
Overview of Progress in Genetics of Psychiatric Disorders
The wealth of data from family, twin, and adoption studies of the major mental disorders exceeds that of all other chronic human diseases. The increased recognition of the role of biologic and genetic vulnerability factors for mental disorders has led to research with increasing methodologic sophistication that has spanned the second half of the 20th century (6–15). There are numerous comprehensive reviews of genetic research on specific disorders of interest as well as on psychiatric genetics in general (16–38).
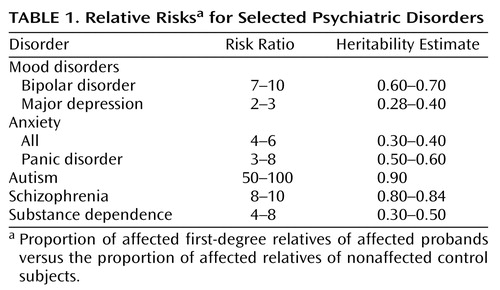
Table 1 presents the relative risks (i.e., proportion affected among first-degree relatives of affected probands versus those of relatives of nonaffected control subjects) derived from controlled family studies of selected psychiatric disorders. The risk ratios comparing the proportion of affected relatives of cases versus control subjects are greatest for autism, bipolar disorder, and schizophrenia; intermediate for substance dependence and subtypes of anxiety, particularly panic; and lowest for major depression. While family studies indicate the degree to which diseases aggregate in families, they alone cannot address the question of genetic versus environmental factors as the source of such aggregation. For this assessment, unusual relationships such as twins, adoptees, or half siblings are required. Estimates of heritability (i.e., the proportion of variance attributable to genetic factors) derived from twin studies, which compare rates of disorders in monozygotic and dizygotic twins, reinforce the notion that genes play a major role in the extent to which mental disorders run in families. The heritability estimates for specific disorders shown in Table 1 are parallel to the risk ratios derived from family studies. Furthermore, adoption and half-sibling studies also support a genetic basis for the observed familial aggregation (17).
An illustration of the application of family, twin, and adoption study research in psychiatry is provided by bipolar disorder, which is one of the most widely studied psychiatric disorders from a genetic perspective (22, 39). Recent reviews reveal that the weighted average rate of bipolar disorder among first-degree relatives of bipolar probands is 5.5%, whereas only 0.6% of the relatives of unaffected control subjects have a history of bipolar illness, yielding a risk ratio of 9.2. More refined estimates are available by age, sex, and comorbid disorders in the proband. These estimates can be used to estimate the risk to relatives of individuals with these conditions.
Adoption studies are the most powerful design to test the relative contributions of genetic and environmental factors to etiology. The aggregate adoption study data on mood disorders reveal a moderate increase in rates of mood disorders among biologic compared with adoptive relatives of adoptees with mood disorders (40). With respect to bipolar disorder, there is little evidence for differential risk among biologic compared with adoptive relatives of adoptees with bipolar disorder. However, the small numbers of bipolar adoptees who have been studied (i.e., less than 50) do not provide an adequate test of genetic and environmental influences (41). The most compelling finding from adoption studies, however, is the dramatic increase in completed suicide among biological relatives of mood disorder probands (42, 43).
Methods for Identifying Genes
Linkage and Linkage Disequilibrium
The traditional approach for locating a disease gene in humans is linkage analysis, which tests the association between DNA polymorphic markers and affected status within families. After linkage is detected with an initial marker, many other markers nearby may also be examined. Markers showing the strongest correlation with disease in families are assumed to be closest to the disease locus.
Linkage analysis uses DNA sequences with high variability (i.e., polymorphisms) in order to increase the power to identify markers that are associated with a disease within families. Historically, different methodological approaches have been applied. Earlier linkage studies employed restriction fragment length polymorphisms (RFLPs) (45), whereas subsequent studies examined short tandem repeat markers, or “microsatellites” (46)—DNA sequences that show considerable variability among people but that have no functional consequences. More recently, linkage and association studies have examined single nucleotide polymorphisms (SNPs) to track diseases in families.
Markers in the candidate region identified by linkage analysis can be used to narrow the location of the disease gene through linkage disequilibrium analysis. Linkage disequilibrium is a population association between two alleles at different loci; it occurs when the same founder mutation exists in a large proportion of affected subjects in the population studied. Usually, the closer the marker is to the disease locus, the greater the proportion of affected subjects who carry the identical allele at the marker (44). However, in measuring the strength of linkage disequilibrium for a given marker, it is also important to select unaffected control subjects from the same population, since an allele shared among affected subjects may also be common in the general population and thus shared by chance rather than due to proximity to the disease locus (44).
For complex human diseases, a simple mode of genetic inheritance is not apparent, and indeed, multiple contributing genetic loci are likely to be involved. Study designs that do not depend on the particular mode of inheritance are required for linkage analysis. Since affected relatives provide most of the information for such analyses, studies that focus on searching for increased sharing of marker alleles above chance expectation among affected relatives may be employed. The simplest of such studies involves affected sibships, where allele sharing in excess of 50% (the expectation when there is no linkage) is sought.
Association Studies
Linkage analysis has not proven successful in identifying genes for most complex diseases, presumably because the effects of the underlying genes are not strong enough to be detected by linkage (47). Therefore, genome-wide association studies have been offered as a more powerful approach. Completion of the human genome project has provided an unprecedented opportunity to identify the effect of gene variants on complex phenotypes, such as psychiatric disorders. Functional genomics technology involving microarrays and proteomics will provide added insights regarding gene function on the cellular level, improving our ability to predict phenotypic effects of genes at the organismic level (44). The recently proposed project (known as the HapMap project) to develop a map of human haplotypes, or blocks of genes that may have been conserved in evolutionary history, has generated considerable enthusiasm for its potential to inform the genetic basis of complex disorders in the general population. There has been considerable discussion regarding the value of studying SNPs with functional significance (47) versus noncoding or evenly spaced SNPs (48). Botstein and Risch (49) have proposed that the initial work employ sequence-based association studies that focus on functional coding regions, rather than a map-based approach (48, 50, 51) that relies solely on the location of haplotypes in order to maximize power and efficiency for the detection of genes for complex human diseases.
Association studies examine candidate genes among affected individuals and unrelated unaffected control subjects. The same limitations that apply to case-control studies of other risk factors must also be considered in genetic case-control studies. The most serious problem in the design of association studies is the failure to select control subjects who are comparable to the cases on all factors except the disease of interest. Failure to equate cases and control subjects may lead to confounding (i.e., a spurious association due to an unmeasured factor that is associated with both the candidate gene and the disease). In genetic case-control studies, the most likely source of confounding is ethnicity because of differential gene and disease frequencies in different ethnic subgroups. Aside from confounding, association studies are particularly prone to false positive findings due to multiple testing without correction and the low prior probability of a gene-disease association (52, 53). The latter problem can be resolved in part by the use of more stringent alpha levels (i.e., false positive error rates) in association studies (47). In addition, there is a strong publication bias against reports of negative association studies (54).
The Importance of Replication
Within the past two decades, linkage analysis has had remarkable success in identifying genes for Mendelian diseases such as cystic fibrosis and Huntington’s disease, among many others. Several of these genes also account for an uncommon subset of generally more common disorders such as breast cancer (BRCA1 and 2), colon cancer (familial adenomatous polyposis), hereditary nonpolyposis colorectal cancer, and Alzheimer’s disease (β-amyloid precursor protein and presenilin-1 and -2).
Despite these successes, linkage studies of complex disorders have been difficult to replicate. A recent review of the linkage findings for 31 complex human diseases based on whole genome screens concluded that the genetic localization of most susceptibility loci is still imprecise and difficult to replicate (55). Increased success in the replication of linkage studies was associated with two study features: an increase in the sample size and ethnic homogeneity of the sample (55). Hirschhorn et al. (54) conducted a similar review of genetic association studies and concluded that few were reproducible. Although these latter reviews do not inspire confidence in the future of these strategies for identifying genes for complex diseases, the studies reviewed still represent the early phases of the application of an extremely powerful technology. Substantial effort will be required to refine phenotypes, identify sources of complexity, and develop new tools and methods to maximize the likelihood of identifying genetic risk factors.
The basis of all scientific research is hypothesis testing and validation of results by independent researchers. Independent replication, typically viewed as the sine qua non for accepting a hypothesis, has become an especially difficult issue in the genetic studies of complex diseases. When a genetic effect is large, most independent researchers can readily obtain similar results with strong levels of statistical significance (44). Most genes for Mendelian disorders have lived up to this expectation. However, when genetic effects are weak and possibly context-dependent (e.g., they may vary by sex, ethnicity, or precision of diagnosis), replication may be particularly difficult, and very large samples may be required before confident conclusions can be drawn.
Application of Linkage and Association to Psychiatric Disorders
With the aforementioned dramatic advances in molecular genetics during the past 20 years, there has been a major shift in the focus of psychiatric genetic investigations from elucidating patterns of familial transmission to localizing genes underlying mental disorders using linkage studies and, more recently, association studies (56, 57). The early successes of linkage studies of Mendelian diseases generated a strong sense of optimism that the same approach would be successful for mental disorders and other complex disorders (58, 59). However, the promise has been unfulfilled to date (21, 24, 56, 57, 60–65). For example, in a recent summary of genome-wide linkage studies of bipolar disorder based on gene scans in a total of 3,538 affected subjects from 1,119 pedigrees reported in 20 samples, Prathikanti and McMahon (66) concluded that no two studies conclusively implicated the same region. The most striking conclusions were that no two studies employed identical ascertainment procedures and that there was substantial diversity in sampling and methods.
Because of increasing skepticism in accepting the findings of linkage and association studies, there has been considerable debate regarding what constitutes acceptable evidence of a true replication. For example, failure to replicate an initial report of linkage between schizophrenia and a marker on chromosome 1q in 22 families (67, 68) was attributed to study design (67) and heterogeneity (69), even though the replication study was conducted on a very large sample of 722 families (70). There have been several recent attempts to standardize the criteria for confirming linkage and association findings.
The ping-pong game between linkage and association claims and disconfirmations has been referred to as a “manic depressive history” by Risch and Botstein (71). It has been particularly difficult for psychiatric clinicians on the front lines to interpret progress in psychiatric genetics because of the inconsistency of findings as well as the difficulty in comprehending the complex methods of molecular biology and the statistical methods of genetic epidemiology. Nevertheless, there are several recent promising findings in psychiatric genetics described in other papers in this issue, although replication remains the cornerstone before such findings can be generally accepted. In addition, the increasing tendency for collaborative efforts on genetics studies within psychiatry may also help to offset some of the aforementioned inconsistencies (35, 70, 72).
Sources of Complexity in Mental Disorders
Two major contributors to the complex patterns of inheritance with regard to psychiatric disorders are 1) the lack of validity of the classification of psychiatric disorders (e.g., phenotypes or observable aspects of diseases) and 2) the complexity of the pathways from genotypes to psychiatric phenotypes.
Lack of Validity of the Classification System
Psychiatric disorder phenotypes, based solely on clinical manifestations without pathognomonic markers, still lack conclusive evidence for the validity of classification and the reliability of measurement (73). This situation is not caused by a lack of attention to classification in psychiatry; in fact, advances in the development of standardized classification and assessment methods have superseded those of most other clinically defined diagnostic entities. The development of structured interviews has enhanced comparability of diagnostic methods within the United States and worldwide. There is now an exciting venture designed to collect information on the prevalence of mental disorders which is using comparable diagnostic tools in more than 20 countries under the auspices of the World Mental Health 2000 Initiative (sponsored by the World Health Organization) (74).
The continued reliance on the descriptive approach as the sole basis for diagnosis in psychiatry can be attributed to the greater complexity of the human brain relative to other human systems. In fact, mental disorders involve the highest level of human functioning. Therefore, the difficulty in classifying human cognition, behavior, and emotion is not unexpected in light of the complex psychological and physiological states underlying mental function, which is the product of the entire human experience in adaptation to the environment (75). Advances in the tools to tap human brain functioning in vivo have led to dramatic advances in knowledge about central nervous system function (76). However, this work is still in its infancy. These advances are likely to yield valuable information for understanding and, hence, classification of mental disorders.
Complex Patterns of Transmission
The application of advances in genomics to mental disorders is still limited by the complexity of the process through which genes exert their influence. There is substantial evidence that a lack of one-to-one correspondence between the genotype and phenotype exists for most of the major mental disorders. Phenomena such as penetrance (i.e., probability of phenotypic expression among individuals with a susceptibility gene), variable expressivity (i.e., variation in clinical expression associated with a particular gene), gene-environment interaction (i.e., expression of genotype only in the presence of particular environmental exposures), pleiotropy (i.e., capacity of genes to manifest several different phenotypes simultaneously), genetic heterogeneity (i.e., different genes leading to indistinguishable phenotypes), and polygenic and oligogenic modes of inheritance (i.e., simultaneous contributions of multiple genes rather than Mendelian single-gene models) are characteristic of the mental disorders, as they are of numerous other complex disorders for which susceptibility genes have been identified (77, 78). Other complicated situations include mitochondrial inheritance, imprinting, and other epigenetic phenomena (79).
The high magnitude of comorbidity and co-aggregation of index disorders with other major psychiatric disorders (i.e., bipolar disorder and alcoholism, major depression and anxiety disorders, schizophrenia and drug dependence), in part induced by the classification system, has been demonstrated in both clinical and community studies (80–85). For example, alcoholism, a well-established complication of bipolar illness, may mask the underlying features of bipolarity, leading to phenotypic misclassification in genetic studies (86). Nonrandom mating is also a common phenomenon in mental disorders that impedes evaluation of patterns of familial transmission (84). Assortative mating is particularly pronounced for substance use disorders for which substance dependence among spouses of substance dependent probands may be as high as 90% (84, 87). These phenomena serve to increase the noise-to-signal ratio in defining the mental disorders for genetic studies. Studies that attempt to identify the impact of these phenomena on phenotypic and endophenotypic expression in individuals and families will bring us closer to understanding the role of the underlying genes on the components of mental disorders.
Future Research to Resolve Sources of Complexity
Use of Endophenotypes for Classification
With plans for the development of the DSM-V underway, it is essential that scientific evidence be used to revise the diagnostic classification system(88). There is abundant research on sources of comorbidity, dimensional classification of disorders, and inclusion of subthreshold diagnostic categories and diagnostic spectra (89–91). As this effort continues, research on the classification of the phenotype for genetic and other biologic studies should increasingly strive for classification that may more closely represent expression of underlying biologic systems.
Phenotypic traits or markers that may represent more direct expressions of underlying genes and the broader disease phenotype have been termed “endophenotypes” (77) (see the review article by Gottesman and Gould in this issue). Studies of the role of genetic factors involved in these systems may be far more informative than studies of the aggregate psychiatric phenotypes. Since endophenotypes should more clearly represent expression of genotypes, it is likely that they will help to unravel the complexity of transmission of the mental disorders. Progress in understanding the pathogenesis of the mental disorders and their component features will enhance identification of endophenotypes and provide a more fertile ground for interaction with basic science. For example, some of the endophenotypes that may underlie mood disorders include circadian rhythm, stress reactivity, and mood, sleep, and appetite regulation (92). Numerous other endophenotypes have also been examined for other disorders, including schizophrenia (77, 93–95), anxiety disorders (65), and attention deficit disorder (96). Some examples of the direct implications of the advances in neuroscience and neuroimaging for phenotypic characterization are described by Thompson et al. (97) and Dolan (75).
Genetic Epidemiology
Applying the tools of genetic epidemiology, particularly when coupled with continued progress in the neurosciences and behavioral sciences, is likely to be one of the most fruitful approaches to resolving sources of complexity in the mental disorders and translating the progress in genomics to the public (98). Figure 1 shows the classic triangle that illustrates the major focus of epidemiologic investigations: the products of the interaction between the host, an infectious or other type of agent, and the environment that promotes the exposure (99). The factors that may be associated with increased risk of human disease are shown under each of the three domains of influence. The field of genetic epidemiology focuses on the role of genetic factors that interact with other domains of risk to enhance vulnerability or protection against disease (100–102). It is quite conceivable that several combinations of these risk factors could produce similar phenotypes in susceptible individuals. The test for epidemiology over the next decades will be to determine the extent to which the tools can be refined to capture these situations.
Sampling
The shift from systematic large-scale family studies to linkage studies in psychiatry has led to the collection of families according to very specific sampling strategies (e.g., many affected relatives, affected sibling pairs, affected relatives on one side of the family only, availability of parents for study, etc.) in order to maximize the power of detecting genes according to the assumed model of familial transmission. Despite the increase in power for detecting genes, these sampling approaches have diminished the generalizability of the study findings and contribute little else to the knowledge base if genes are not discovered. As we learn more about the complexity of genetic risk factors, it may be advisable in the future to collect both families and control subjects from representative samples of the population in order to enable estimation of population risk parameters, enhance generalizability, and examine the specificity of endophenotypic transmission.
Study Designs
Epidemiologic studies generally proceed from retrospective case-control studies to develop specific hypotheses that can be addressed in prospective cohort studies in order to demonstrate causality. The major goal of analytic epidemiology is to identify risk and protective factors and their causal links to disease, with the ultimate goal of disease prevention. Genetic epidemiology employs traditional epidemiologic study designs to identify explanatory factors for aggregation in groups of relatives ranging from twins to migrant cohorts. The tools of genetic epidemiology will be employed in the era of genomics to derive estimates of the population distribution of disease genes, to test modes of disease transmission in systematic samples that are representative of the population, and to identify sources of gene-environment interactions for diseases. Since epidemiology has developed sophisticated designs and analytic methods for identifying disease risk factors, these methods can now be extended to include both genes and environmental factors as gene identification proceeds.
In general, study designs in genetic epidemiology either control for genetic background while letting the environment vary (e.g., migrant studies, half siblings, separated twins) or control for the environment while allowing variance in the genetic background (e.g., siblings, twins, adoptees-nonbiological siblings). Because each of the study designs has both strengths and limitations, it is important to evaluate aggregaate evidence from multiple approaches to yield conclusive evidence regarding the role of genetic and environmental risk factors. Over the next decades, it will be important to identify and evaluate the effects of specific environmental factors on disease outcomes and to refine measurement of environmental exposures to evaluate specificity of effects.
Migrant studies are perhaps the most powerful study design to identify environmental and cultural risk factors. One of the earliest controlled migrant studies evaluated rates of psychosis among Norwegian immigrants to Minnesota compared with native Minnesotans and native Norwegians (103). The higher rate of psychosis among the immigrants than in both the native Minnesotans and Norwegians was attributed to greater susceptibility to psychosis among the migrants who left Norway. It was found that migration selection bias was the major explanatory factor rather than an environmental exposure in the new culture (103).
Another powerful study design is the nested case-control study built on an established cohort. Prospective cohort studies are also valuable sources of diagnostic stability, causal associations between risk factors and disease, and developmental aspects of psychiatric disorders. Langholz et al. (104) described some of the world’s prospective cohort studies that may serve as a basis for studies of gene-disease associations or gene-environment interactions. Finally, the half-sibling approach may eventually replace the adoption paradigm to investigate genetic and environmental effects because of the recent trends toward selective adoption and the diminishing frequency of adoptions in the United States and in numerous other countries (i.e., maternal selection of adoptive parents and continued contact with biological mothers).
Population-Based Studies
The importance of epidemiology to the future of genetics has been described by numerous geneticists and epidemiologists who conclude that the best strategy for gene identification will ultimately involve large epidemiologic studies from diverse populations (39, 44, 47, 98, 105–108). It is likely that population-based association studies will assume increasing importance in translating the products of genomics to public health (47). The term “human genome epidemiology” was coined by Khoury et al. (108) to denote the emerging field that employs systematic applications of epidemiologic methods in population-based studies of the impact of human genetic variation on health and disease.
There are several reasons that population-based studies will be critical to the future of genetics. First, the prevalence of newly identified polymorphisms, whether SNPs or other variants, especially in particular population subgroups, is not known. Second, current knowledge of genes as risk factors is based nearly exclusively on clinical and nonsystematic samples. Hence, the significance of the susceptibility alleles that have been identified for cancer, heart disease, diabetes, and so forth is unknown in the population at large. In order to provide accurate risk estimates, the next stage of research needs to move beyond samples identified through affected individuals to the population in order to obtain estimates of the risk of specific polymorphisms for the population as a whole. Third, identification of risk profiles will require very large samples to assess the significance of vulnerability genes with relatively low expected population frequencies. Fourth, similar to the role of epidemiology in quantifying risk associated with traditional disease risk factors, applications of human genome epidemiology can provide information on the specificity, sensitivity, and impact of genetic tests to inform science and the individual (107).
Because genetic polymorphisms involved in complex diseases are likely to be nondeterministic (i.e., the marker neither predicts disease nor nondisease with certainty), traditional epidemiologic risk factor designs can be used to estimate their impact (101). As epidemiologists add genes to their risk equations, it is likely that the contradictory findings from studies that have generally employed solely environmental risk factors, such as diet, smoking, alcohol use, etc., will be resolved. Likewise, the studies that seek solely to identify genes will also continue to be inconsistent without considering the effects of nongenetic biologic parameters as well as environmental factors that contribute to the diseases of interest.
There are several types of risk estimates that are used in public health. The most common is relative risk, the magnitude of the association between an exposure and disease. It is independent of the prevalence of the exposure. The absolute risk is the overall probability of developing a disease in a particular population (99). The population attributable risk relates to the risk of a disease in a total population (exposed and unexposed) and indicates the amount the disease can be reduced in a population if an exposure were eliminated. The population attributable risk depends on the prevalence of the exposure, or in the case of genes, the gene frequency. Genetic attributable risk would indicate the proportion of a particular disease that may be attributed to a particular genetic locus.
Figure 2 illustrates the known genetic and environmental risk factors for Alzheimer’s disease (109). The orange areas on the left represent the roles of deterministic genes (β-amyloid precursor, presenilin-1 and -2) and the susceptibility gene apolipoprotein-E ε4 (APOE-ε4) (110). The blue areas on the right indicate environmental risk and protective factors, respectively (111–113). Individuals with mutations in deterministic genes appear to have nearly a 100% chance (i.e., fully penetrant) for the development of Alzheimer’s disease. Likewise, the relative risk of these genes would also be quite high. In contrast, because these mutations are presumed to be very rare in the population, the population attributable risk is quite low, meaning that were these mutations to be eliminated from the population, there would be little impact on the prevalence of Alzheimer’s disease.
APOE ε4 The APOE ε4 allele has been shown to increase the risk of Alzheimer’s disease in a dose-dependent fashion. Using data from a large multiethnic sample collected by more than 40 research teams, Farrer et al. (114) reported a 2.6–3.2 greater odds of Alzheimer’s disease among those with one copy, and 14.9 odds of Alzheimer’s disease among those with two copies of the APOE ε4 allele. Moreover, there was a significant protective effect among those with the ε2/ ε3 genotype. As opposed to the deterministic mutations, the APOE ε4 allele has a very high population attributable risk because of its high frequency in the population.
Identification of Environmental Factors
The identification of gene-environment interactions will be one of the most important future goals of genetic epidemiology. Newman et al. (115) credit the synergy between genetics and epidemiology for elucidating the initial gene findings as well as for the subsequent identification of other susceptibility alleles and the environmental factors that may influence the risk of breast cancer in susceptible persons. Study designs and statistical methods should focus increasingly on gene-environment interaction (116–122). Evidence is emerging that gene-environment interaction underlies many of the complex human diseases. Some examples include inborn errors of metabolism, individual variation in response to drugs (123), substance use disorders (124, 125), and the protective influence of a deletion in the CCRS gene on exposure to HIV (79, 126).
With respect to mental disorders, recent reviews of prospective studies that evaluated environmental risk factors for the common mental disorders ascertained in population-based studies yielded few specific environmental factors that could be etiologically linked with any of the major mental disorders (127). However, one promising exception is the increasing evidence from genetic epidemiologic studies that environmental exposures including pre- and perinatal factors, such as viral agents, may enhance the risk of schizophrenia (128). Other informative study designs for identifying gene-environment interactions include migrant studies and genetic case-control studies in which the cases may be defined by a genetic susceptibility marker.
Future research designed to identify environmental factors that operate either specifically or nonspecifically on those with susceptibility to mental disorders may provide an important opportunity for prevention and intervention, once susceptibility genes have been identified. The recent advances in understanding the bidirectional communication of neural systems and experience (76) provide an ideal opportunity to apply genetic epidemiologic methods such as case-control and prospective cohort studies. Increased knowledge of the developmental pathways of emotion, cognition, and behavior will expand our ability to identify specific environmental factors such as infection, poor diet, prenatal environment, and early life experiences that interact with the genetic architecture of mood regulation and cognition (129).
Impact of Genomics on Psychiatric Science and Practice
Will genomics lead to changes in medical practice and transform psychiatric genetics?
In a recent summary of implications of the genome for medical practice, Varmus (130) concluded that despite the journalistic hyperbole, the sequencing of the human genome is unlikely to lead to either a radical transformation of medical practice or even to an information-based science that can predict with certainty future diseases and effective treatment interventions. Although this skepticism may be somewhat extreme, it is clear that progress in genomics has far outweighed advances in our understanding of psychiatric phenotypes and the complexity underlying their etiology, as well as our current armamentarium to identify genetic and environmental risk factors. Therefore, despite the extraordinary opportunity for understanding disease pathogenesis afforded by the technical advances and availability of rapidly expanding genetic databases, it is unlikely that we will soon experience the light speed progress of genomics in understanding, treating, or preventing major mental disorders. The chasm between genetic information and clinical utility should gradually close as we develop new methods and tools in human genetic and clinical research to maximize the knowledge afforded by the exciting advances in genomics.
Increased integration of advances in neuroscience (131) and genomics (see the series of papers in the New England Journal of Medicine such as the primer by Guttmacher and Collins [79]), along with information from population-based studies and longitudinal cohorts, innovations in our conceptualizations of the mental disorders, and the identification of specific risk and protective factors, will lead to more informed intervention strategies in psychiatry. As we learn more about the role of genes as risk factors, rather than as the chief causes of common human diseases, it will be essential to provide accurate risk estimation and to inform the public of the need for population-based integrated data on genetic, biologic, and environmental risk factors.
The goal of genomics research is ultimately prevention, the cornerstone of public health. Gaining understanding of the significance of genetic risk factors and learning proper interpretation of their meaning for patients and their families will ultimately become part of clinical practice, and clinicians will be involved more than ever in helping patients to comprehend the meaning and potential impact of genetic risk for nonpsychiatric disorders as well. As our knowledge on the role of genes for mental disorders advances, it will be incumbent upon clinicians to become familiar with knowledge gleaned from genetic epidemiologic and genomics research. However, the deterministic view of the father in this father-son pair in Figure 3 is unlikely to be scientifically justified anytime soon. In the meanwhile, recurrence risk estimates from family studies constitute the best available knowledge on which to predict the risk of the development of mental disorders.
 |
From the NIMH Mood and Anxiety Disorders Program; the Department of Genetics, Stanford University School of Medicine, Stanford, CA, and the Division of Research, Kaiser Permanente, Oakland, CA Address reprint requests to Dr. Merikangas, Section on Developmental Genetic Epidemiology, Mood and Anxiety Disorders Program, National Institute of Mental Health, National Institutes of Health, Department of Health and Human Services, 15K North Dr., MSC 2670, Bethesda, MD 20892; [email protected] (e-mail).

Figure 1. The Epidemiologic Triangle

Figure 2. Genetic and Environmental Factors in Alzheimer’s Disease

Figure 3. The Perils of Genetic Determinism?
1. Kallmann FJ: Review of psychiatric progress 1953. Am J Psychiatry 1954; 110:489-492Link, Google Scholar
2. Kallmann FJ, Jarvik LF: Individual differences in constitution and genetic background, in Aging and the Individual. Edited by Birren JE. Chicago, University of Chicago Press, 1959Google Scholar
3. Stenstedt A: A study in manic-depressive psychosis: clinical, social, and genetic investigations. Acta Psychiatr Neurol Scand 1952; 42:398-409Google Scholar
4. Slater E: The incidence of mental disorder. Annals of Eugenics 1935; 6:172Crossref, Google Scholar
5. Böök JA: A genetic and neuropsychiatric investigation of a north-Swedish population with special regard to schizophrenia and mental deficiency. Acta Genet Stat Med 1953; 4:70-84Google Scholar
6. Reich T, Clayton PJ, Winokur G: Family history studies, V: the genetics of mania. Am J Psychiatry 1969; 125:1358-1369Link, Google Scholar
7. Tsuang M, Dempsey G, Dvoredsky A, Strauss A: A family history study of schizoaffective disorder. Biol Psychiatry 1977; 12:331-338Medline, Google Scholar
8. Winokur G, Tsuang MT, Crowe RR: The Iowa 500: affective disorder in relatives of manic and depressed patients. Am J Psychiatry 1982; 139:209-212Link, Google Scholar
9. Andreasen NC, Rice J, Endicott J, Coryell W, Grove WM, Reich T: Familial rates of affective disorder: a report from the National Institute of Mental Health Collaborative Study. Arch Gen Psychiatry 1987; 44:461-469; correction, 1988; 45:776Google Scholar
10. Gershon ES, Hamovit J, Guroff JJ: A family study of schizoaffective, bipolar I, bipolar II, unipolar, and normal control probands. Arch Gen Psychiatry 1982; 39:1157-1167Crossref, Medline, Google Scholar
11. Weissman MM, Kidd KK, Prusoff BA: Variability in rates of affective disorders in relatives of depressed and normal probands. Arch Gen Psychiatry 1982; 39:1397-1403Crossref, Medline, Google Scholar
12. Kendler KS: Twin studies of psychiatric illness: an update. Arch Gen Psychiatry 2001; 58:1005-1014Crossref, Medline, Google Scholar
13. Kety SS, Rosenthal D, Wender PH, Schulsinger F: The types of prevalence of mental illness in the biological and adoptive families of adopted schizophrenics, in The Transmission of Schizophrenia. Edited by Rosenthal D, Kety SS. Oxford, UK, Pergamon Press, 1968Google Scholar
14. Rosenthal D: Some factors associated with concordance and discordance with respect to schizophrenia in monozygotic twins. J Nerv Ment Dis 1959; 129:1-10Crossref, Medline, Google Scholar
15. Weissman MM, Gershon ES, Kidd KK, Prusoff BA, Leckman JF, Dibble E, Hamovit JH, Thompson WD, Pauls DL, Guroff JJ: Psychiatric disorder in relatives of probands with affective disorders: the Yale University-NIMH Collaborative Family Study. Arch Gen Psychiatry 1984; 41:13-21Crossref, Medline, Google Scholar
16. Plomin R, Defries JC, McClearn GE, McGuffin F: Behavioral Genetics. London, WH Freeman and Company, 2000Google Scholar
17. Merikangas KR, Swendsen JD: Genetic epidemiology of psychiatric disorders. Epidemiol Rev 1996; 19:1-12Google Scholar
18. McGuffin P, Owen MJ, Gottesman II: Psychiatric Genetics and Genomics. Oxford, UK, Oxford University Press, 2002Google Scholar
19. Moldin SO: Summary of research—appendix to the report of the NIMH’s Genetics Workgroup. Biol Psychiatry 1999; 45:573-602Crossref, Google Scholar
20. McGuffin P, Asherson P, Owen M, Farmer A: The strength of the genetic effect: is there room for an environmental influence in the aetiology of schizophrenia? Br J Psychiatry 1994; 164:593-599Crossref, Medline, Google Scholar
21. Craddock N, Jones I: Molecular genetics of bipolar disorder. Br J Psychiatry Suppl 2001; 41:S128-S133Google Scholar
22. Sullivan PF, Neale MC, Kendler KS: Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000; 157:1552-1562Link, Google Scholar
23. Kendler KS, Neale MC, Kessler RC: Panic disorder in women: a population-based twin study. Psychol Med 1993; 23:397-406Crossref, Medline, Google Scholar
24. van den Heuvel OA, van de Wetering BJ, Veltman DJ, Pauls DL: Genetic studies of panic disorder: a review. J Clin Psychiatry 2000; 61:756-766Crossref, Medline, Google Scholar
25. Smoller JW, Finn C, White C: The genetics of anxiety disorders: an overview. Psychiatr Annals 2000; 30:745-753Crossref, Google Scholar
26. Nestadt G, Samuels J, Riddle M, Bienvenu OJI, Liang K-Y, LaBuda M, Walkup J, Grados M, Hoehn-Saric R: A family study of obsessive-compulsive disorder. Arch Gen Psychiatry 2000; 57:358-363Crossref, Medline, Google Scholar
27. Sullivan PF, Kendler KS: Typology of common psychiatric syndromes: an empirical study. Br J Psychiatry 1998; 173:312-319Crossref, Medline, Google Scholar
28. Kendler KS, Prescott CA: Cannabis use, abuse, and dependence in a population-based sample of female twins. Am J Psychiatry 1998; 155:1016-1022Link, Google Scholar
29. Kendler K, Prescott C: Cocaine use, abuse and dependence in a population-based sample of female twins. Br J Psychiatry 1998; 173:345-350Crossref, Medline, Google Scholar
30. Merikangas KR: Genetic epidemiology of substance-use disorders, in Textbook of Biological Psychiatry. Edited by D’haenen, Den Boer J, Willner P. New York, John Wiley & Sons, 2002, pp 537-546Google Scholar
31. Uhl GR, Liu QR, Naiman D: Substance abuse vulnerability loci: converging genome scanning data. Trends Genet 2002; 18:420-425Crossref, Medline, Google Scholar
32. Rice F, Harold G, Thapar A: The genetic aetiology of childhood depression: a review. J Child Psychol Psychiatry 2002; 43:65-79Crossref, Medline, Google Scholar
33. Szatmari P, Jones MB, Zwaigenbaum L, MacLean JE: Genetics of autism: overview and new directions. J Autism Dev Disord 1998; 28:351-368Crossref, Medline, Google Scholar
34. Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, Nguyen L, Yang J, Harper C, Thorpe D, Vermeer S, Young H, Hebert J, Lin A, Ferguson J, Chiotti C, Wiese-Slater S, Rogers T, Salmon B, Nicholas P, Myers RM, et al: A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 1999; 65:493-507Crossref, Medline, Google Scholar
35. International Molecular Genetic Study of Autism Consortium: A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet 1998; 7:571-578Crossref, Medline, Google Scholar
36. Thapar A, Holmes J, Poulton K, Harrington R: Genetic basis of attention deficit and hyperactivity. Br J Psychiatry 1999; 174:105-111Crossref, Medline, Google Scholar
37. Thapar A, Scourfield J: Childhood disorders, in Psychiatric Genetics and Genomics. Edited by McGuffin P, Owen MJ, Gottesman II. Oxford, UK, Oxford University Press, 2002, pp 147-180Google Scholar
38. Rutter M, Silberg J, O’Connor T, Simonoff E: Genetics and child psychiatry, II: empirical research findings. J Child Psychol Psychiatry 1999; 40:19-55Crossref, Medline, Google Scholar
39. Merikangas KR, Chakravarti A, Moldin SO, Araj H, Blangero J, Burmeister M, Crabbe JCJ, DePaulo JRJ, Foulks E, Freimer NB, Koretz DS, Lichtenstein W, Mignot E, Reiss AL, Risch NJ, Takahashi J (Workgroup on Genetics for NIMH Strategic Plan for Mood Disorders): Future of genetics of mood disorders research. Biol Psychiatry 2002; 52:457-477Crossref, Medline, Google Scholar
40. Tsuang MT, Faraone SV: The Genetics of Mood Disorders. Baltimore, Johns Hopkins University Press, 1990Google Scholar
41. Goodwin FK, Jamison KR: Manic-Depressive Illness. New York, Oxford University Press, 1990Google Scholar
42. Wender PH, Kety SS, Rosenthal D, Schulsinger F, Ortmann J, Lunde I: Psychiatric disorders in the biological and adoptive families of adopted individuals with affective disorders. Arch Gen Psychiatry 1986; 43:923-929Crossref, Medline, Google Scholar
43. Mendlewicz J, Rainer JD: Adoption study supporting genetic transmission in manic-depressive illness. Nature 1977; 268:326-329Crossref, Google Scholar
44. Risch NJ: Searching for genetic determinants in the new millennium. Nature 2000; 405:847-856Crossref, Medline, Google Scholar
45. Botstein D, White RL, Skolnick M, Davis RW: Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 1980; 32:314-331Medline, Google Scholar
46. Weber JL, May PE: Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 1989; 44:388-396Medline, Google Scholar
47. Risch N, Merikangas KR: The future of genetic studies of complex human diseases. Science 1996; 273:1516-1517Crossref, Medline, Google Scholar
48. Collins FS, Guyer MS, Charkravarti A: Variations on a theme: cataloging human DNA sequence variation. Science 1997; 278:1580-1581Crossref, Medline, Google Scholar
49. Botstein D, Risch N: Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet Suppl 2003; 33Google Scholar
50. Patil N: Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 2001; 294:1719-1723Crossref, Medline, Google Scholar
51. Gabriel SB: The structure of haplotype blocks in the human genome. Science 2002; 296:2225-2228Crossref, Medline, Google Scholar
52. Wacholder S, Rothman N, Caporaso N: Population stratification in epidemiologic studies of common genetic variants and cancer: quantification of bias. J Natl Cancer Inst 2000; 92:1151-1158Crossref, Medline, Google Scholar
53. Wacholder S, Garcia-Closas M, Rothman N: Study of genes and environmental factors in complex diseases (letter). Lancet 2002; 359:1155Crossref, Medline, Google Scholar
54. Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K: A comprehensive review of genetic association studies. Genet Med 2002; 4:45-61Crossref, Medline, Google Scholar
55. Altmuller J, Palmer LJ, Fischer G, Scherb H, Wjst M: Genomewide scans of complex human diseases: true linkage is hard to find. Am J Hum Genet 2001; 69:936-950Crossref, Medline, Google Scholar
56. McInnes LA, Freimer NB: Mapping genes for psychiatric disorders and behavioral traits. Curr Opin Genet Dev 1995; 5:376-381Crossref, Medline, Google Scholar
57. DeLisi LE, Craddock NJ, Detera-Wadleigh S, Foroud T, Gejman P, Kennedy JL, Lendon C, Macciardi F, McKeon P, Mynett-Johnson L, Nurnberger JI Jr, Paterson A, Schwab S, Van Broeckhoven C, Wildenauer D, Crow TJ: Update on chromosomal locations for psychiatric disorders: report of the interim meeting of chromosome workshop chairpersons from the VIIth World Congress of Psychiatric Genetics, Monterey, California, October 14-18, 1999. Am J Med Genet 2000; 96:434-449Crossref, Medline, Google Scholar
58. Risch N: Searching for genes in complex diseases: lessons from systemic lupus erythematosus. J Clin Invest 2000; 105:1503-1506Crossref, Medline, Google Scholar
59. Glazier AM, Nadeau JH, Aitman TJ: Finding genes that underlie complex traits. Science 2002; 298:2345-2349Crossref, Medline, Google Scholar
60. Moises HW, Yang L, Kristbjarnarson H, Wiese C, Byerley W, Macciardi F, Arolt V, Blackwood D, Liu X, Sjogren B, et al: An international two-stage genome-wide search for schizophrenia susceptibility genes. Nat Genet 1995; 11:321-324Crossref, Medline, Google Scholar
61. Faraone SV, Matise T, Svrakic D, Pepple J, Malaspina D, Suarez B, Hampe C, Zambuto CT, Schmitt K, Meyer J, Markel P, Lee H, Harkavy Friedman J, Kaufmann C, Cloninger CR, Tsuang MT: Genome scan of European-American schizophrenia pedigrees: results of the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet 1998; 81:290-295Crossref, Medline, Google Scholar
62. O’Donovan MC, Owen MJ: Candidate-gene association studies of schizophrenia. Am J Hum Genet 1999; 65:587-592Crossref, Medline, Google Scholar
63. Riley BP, McGuffin P: Linkage and associated studies of schizophrenia. Am J Med Genet 2000; 97:23-44Crossref, Medline, Google Scholar
64. Sklar P: Linkage analysis in psychiatric disorders: the emerging picture. Annu Rev Genomics Hum Genet 2002; 3:371-413Crossref, Medline, Google Scholar
65. Smoller JW, Tsuang MT: Panic and phobic anxiety: defining phenotypes for genetic science. Am J Psychiatry 1998; 155:1152-1162Link, Google Scholar
66. Prathikanti S, McMahon FJ: Genome scans for susceptibility genes in bipolar affective disorder. Ann Med 2001; 33:257-262Crossref, Medline, Google Scholar
67. Bassett AS, Chow EW, Vieland VJ, Brzustowicz L: Is schizophrenia linked to chromosome 1q? (letter). Science 2002; 298:2277Crossref, Google Scholar
68. Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS: Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science 2000; 288:678-682Crossref, Medline, Google Scholar
69. Macgregor S, Visscher PM, Knott S, Porteous D, Muir W, Millar K, Blackwood D: Is schizophrenia linked to chromosome 1q? (letter). Science 2002; 298:2277Crossref, Google Scholar
70. Levinson DF, Holmans PA, Laurent C, Riley B, Pulver AE, Gejman PV, Schwab SG, Williams NM, Owen MJ, Wildenauer DB, Sanders AR, Nestadt G, Mowry BJ, Wormley B, Bauche S, Soubigou S, Ribble R, Nertney DA, Liang KY, Martinolich L, Maier W, Norton N, Williams H, Albus M, Carpenter EB, DeMarchi N, Ewen-White KR, Walsh D, Jay M, Deleuze JF, O’Neill FA, Papadimitriou G, Weilbaecher A, Lerer B, O’Donovan MC, Dikeos D, Silverman JM, Kendler KS, Mallet J, Crowe RR, Walters M: No major schizophrenia locus detected on chromosome 1q in a large multicenter sample. Science 2002; 296:739-741Crossref, Medline, Google Scholar
71. Risch N, Botstein DA: A manic depressive history. Nat Genet 1996; 12:351-353Crossref, Medline, Google Scholar
72. Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G: A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. Proc Natl Acad Sci USA 1999; 96:5604-5609Crossref, Medline, Google Scholar
73. Kendell RE: Clinical validity. Psychol Med 1989; 19:45-55Crossref, Medline, Google Scholar
74. Kessler RC, Üstün TB: The World Health Organization World Mental Health 2000 Initiative. Hosp Management Int 2000:195-196Google Scholar
75. Dolan RJ: Emotion, cognition, and behavior. Science 2002; 298:1191-1194Crossref, Medline, Google Scholar
76. McKhann GM: Neurology: then, now, and in the future. Arch Neurol 2002; 59:1369-1373Medline, Google Scholar
77. Gottesman I, Shields J: Schizophrenia and Genetics: A Twin Study Vantage Point. New York, Academic Press, 1972Google Scholar
78. Risch N: Linkage strategies for genetically complex traits, I: multilocus models. Am J Hum Genet 1990; 46:222-228Medline, Google Scholar
79. Guttmacher AE, Collins FS: Genomic medicine—a primer. N Engl J Med 2002; 347:1512-1520Crossref, Medline, Google Scholar
80. Maier W, Merikangas KR: Co-occurrence and cotransmission of affective disorders and alcoholism in families. Br J Psychiatry Suppl 1996; 168:93-100Google Scholar
81. Merikangas KR, Angst J, Eaton W, Canino G, Rubio-Stipec M, Wacker H, Wittchen HU, Andrade L, Essau C, Whitaker A, Kraemer H, Robins LN, Kupfer DJ: Comorbidity and boundaries of affective disorders with anxiety disorders and substance misuse: results of an international task force. Br J Psychiatry Suppl 1996; 30:58-67Medline, Google Scholar
82. Merikangas KR, Stevens DE, Fenton B, Stolar M, O’Malley S, Woods SW, Risch N: Comorbidity and familial aggregation of alcoholism and anxiety disorders. Psychol Med 1998; 28:773-788Crossref, Medline, Google Scholar
83. Maier W, Lichtermann D, Minges J, Delmo C, Heun R: The relationship between bipolar disorder and alcoholism: a controlled family study. Psychol Med 1995; 25:787-796Crossref, Medline, Google Scholar
84. Merikangas KR: Assortative mating for psychiatric disorders and psychological traits. Arch Gen Psychiatry 1982; 39:1173-1180Crossref, Medline, Google Scholar
85. Galbaud du Fort G, Bland RC, Newman SC, Boothroyd LJ: Spouse similarity for lifetime psychiatric history in the general population. Psychol Med 1998; 28:789-802Crossref, Medline, Google Scholar
86. Merikangas KR, Gelernter CS: Comorbidity for alcoholism and depression. Psychiatr Clin North Am 1990; 13:613-632Crossref, Medline, Google Scholar
87. Merikangas KR, Rounsaville BJ, Prusoff BA: Familial factors in vulnerability to substance abuse, in Vulnerability to Drug Abuse. Edited by Glantz MD, Pickens RW. Washington, DC, American Psychological Association, 1992, pp 75-98Google Scholar
88. Kupfer DJ, First MB, Regier DA: A Research Agenda for DSM-V. Washington, DC, APA, 2002Google Scholar
89. Angst J, Merikangas KR, Preisig M: Subthreshold syndromes of depression and anxiety in the community. J Clin Psychiatry 1997; 58(suppl 8):6-10Google Scholar
90. Judd LL, Akiskal HS, Maser JD, Zeller PJ, Endicott J, Coryell W, Paulus MP, Kunovac JL, Leon AC, Mueller TI, Rice JA, Keller MB: A prospective 12-year study of subsyndromal and syndromal depressive symptoms in unipolar major depressive disorders. Arch Gen Psychiatry 1998; 55:694-700Crossref, Medline, Google Scholar
91. Angst J, Merikangas KR: The depressive spectrum: diagnostic classification and course. J Affect Disord 1997; 45:31-40Crossref, Medline, Google Scholar
92. Lenox RH, Gould TD, Manji HK: Endophenotypes in bipolar disorder. Am J Med Genet 2002; 114:391-406Crossref, Medline, Google Scholar
93. Tsuang MT, Faraone SV, Lyons MJ: Identification of the phenotype in psychiatric genetics. Eur Arch Psychiatry Clin Neurosci 1993; 243:131-142Crossref, Medline, Google Scholar
94. Faraone SV, Kremen WS, Lyons MJ, Pepple JR, Seidman LJ, Tsuang MT: Diagnostic accuracy and linkage analysis: how useful are schizophrenia spectrum phenotypes? Am J Psychiatry 1995; 152:1286-1290Link, Google Scholar
95. Lichtermann D, Karbe E, Maier W: The genetic epidemiology of schizophrenia and of schizophrenia spectrum disorders. Eur Arch Psychiatry Clin Neurosci 2000; 250:304-310Crossref, Medline, Google Scholar
96. Castellanos FX, Tannock R: Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci 2002; 3:617-628Crossref, Medline, Google Scholar
97. Thompson PM, Rapoport JL, Cannon TD, Toga AW: Imaging the brain as schizophrenia develops: dynamic and genetic brain maps. Primary Psychiatry, Nov 2002, pp 40-44Google Scholar
98. Merikangas KR: Genetic epidemiology: bringing genetics to the population—the NAPE Lecture 2001. Acta Psychiatr Scand 2002; 105:3-13Crossref, Medline, Google Scholar
99. Gordis L: Epidemiology. Philadelphia, WB Saunders, 2000Google Scholar
100. Ellsworth DL, Manolio TA: The emerging importance of genetics in epidemiologic research, II: issues in study design and gene mapping. Ann Epidemiol 1999; 9:75-90Crossref, Medline, Google Scholar
101. Ellsworth DL, Manolio TA: The emerging importance of genetics in epidemiologic research, III: bioinformatics and statistical genetic methods. Ann Epidemiol 1999; 9:207-224Crossref, Medline, Google Scholar
102. Khoury MJ, Beaty TH, Cohen BH: Fundamentals of Genetic Epidemiology. New York, Oxford University Press, 1993Google Scholar
103. Ödegaard Ö: Emigration and Insanity: A Study of Mental Disorders Among the Norwegian Born Population of Minnesota. Copenhagen, Levin & Munksgaards, 1932Google Scholar
104. Langholz B, Rothman N, Wacholder S, Thomas DC: Cohort studies for characterizing measured genes. J Natl Cancer Inst Monogr 1999; 26:39-42Crossref, Medline, Google Scholar
105. Khoury MJ, Yang Q: The future of genetic studies of complex human disease: an epidemiologic perspective. Epidemiology 1998; 9:350-354Crossref, Medline, Google Scholar
106. Thomas DC: Genetic epidemiology with a capital “E.” Genet Epidemiol 2000; 19:289-300Crossref, Medline, Google Scholar
107. Yang Q, Khoury MJ, Coughlin SC, Sun F, Flanders WD: On the use of population-based registries in the clinical validation of genetic tests for disease susceptibility. Genet Med 2000; 2:186-192Crossref, Medline, Google Scholar
108. Khoury MJ, McCabe LL, McCabe ER: Population screening in the age of genomic medicine. N Engl J Med 2003; 348:50-58Crossref, Medline, Google Scholar
109. Slooter A, van Duijn C: Genetic epidemiology of Alzheimer disease. Epidemiol Rev 1997; 19:107-119Crossref, Medline, Google Scholar
110. Tol J, Roks G, Slooter AJ, van Duijn CM: Genetic and environmental factors in Alzheimer’s disease. Rev Neurol (Paris) 1999; 155(suppl 4):S10-S16Google Scholar
111. Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A: Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. Br Med J 2001; 322:1447-1451Crossref, Medline, Google Scholar
112. Munoz DG, Feldman H: Causes of Alzheimer’s disease. Can Med Assoc J 2000; 162:65-72Google Scholar
113. Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Iivonen S, Mannermaa A, Tuomilehto J, Nissinen A, Soininen H: Apolipoprotein E epsilon 4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 2002; 137:149-155Crossref, Medline, Google Scholar
114. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (APOE and Alzheimer Disease Meta Analysis Consortium): Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 1997; 278:1349-1356Crossref, Medline, Google Scholar
115. Newman B, Millikan RC, King M: Genetic epidemiology of breast and ovarian cancers. Epidemiol Rev 1997; 19:69-79Crossref, Medline, Google Scholar
116. Ottman R: An epidemiologic approach to gene-environment interaction. Genet Epidemiol 1990; 7:177-185Crossref, Medline, Google Scholar
117. Smith PG, Day NE: The design of case-control studies: the influence of confounding and interaction effects. Int J Epidemiol 1984; 13:356-365Crossref, Medline, Google Scholar
118. Hwang SJ, Beaty TH, Liang KY, Coresh J, Khoury MJ: Minimum sample size estimation to detect gene-environment interaction in case-control designs. Am J Epidemiol 1994; 140:1029-1037Medline, Google Scholar
119. Khoury MJ: Genetic epidemiology, in Modern Epidemiology, 2nd ed. Edited by Rothman K, Greenland S. Philadelphia, Lippincott-Raven, 1997Google Scholar
120. Yang Q, Khoury MJ: Evolving methods in genetic epidemiology, III: gene-environment interaction in epidemiologic research. Epidemiol Rev 1997; 19:33-43Crossref, Medline, Google Scholar
121. Foppa I, Spiegelman D: Power and sample size calculations for case-control studies of gene-environment interactions with a polytomous exposure variable. Am J Epidemiol 1997; 146:596-604Crossref, Medline, Google Scholar
122. Garcia-Closas M, Lubin JH: Power and sample size calculations in case-control studies of gene-environment interactions: comments on different approaches. Am J Epidemiol 1999; 149:689-692Crossref, Medline, Google Scholar
123. Nebert DW, Ingelman-Sundberg M, Daly AK: Genetic epidemiology of environmental toxicity and cancer susceptibility: human allelic polymorphisms in drug-metabolizing enzyme genes, their functional importance, and nomenclature issues. Drug Metab Rev 1999; 31:467-487Crossref, Medline, Google Scholar
124. Heath AC, Whitfield JB, Madden PA, Bucholz KK, Dinwiddie SH, Slutske WS, Bierut LJ, Statham DB, Martin NG: Towards a molecular epidemiology of alcohol dependence: analysing the interplay of genetic and environmental risk factors. Br J Psychiatry Suppl 2001; 40:S33-S40Google Scholar
125. Dick DM, Rose RJ, Viken RJ, Kaprio J, Koskenvuo M: Exploring gene-environment interactions: socioregional moderation of alcohol use. J Abnorm Psychol 2001; 110:625-632Crossref, Medline, Google Scholar
126. Michael NL: Host genetic influences on HIV-1 pathogenesis. Curr Opin Immunol 1999; 11:466-474Crossref, Medline, Google Scholar
127. Eaton WW, Addington AM, Bass J, Forman V, Gilbert S, Hayden K, Mielke M: Risk factors for major mental disorders: a review of the epidemiologic literature. Baltimore, Johns Hopkins University, Bloomberg School of Mental Health, Department of Mental Hygiene, Oct 2002. http://www.jhu.edu/~janthony/share/Envirome/Envirome-III-b.pdfGoogle Scholar
128. Gottesman II, Erlenmeyer-Kimling L: Family and twin strategies as a head start in defining prodromes and endophenotypes for hypothetical early-interventions in schizophrenia. Schizophr Res 2001; 51:93-102Crossref, Medline, Google Scholar
129. Meaney MJ: Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci 2001; 24:1161-1192Crossref, Medline, Google Scholar
130. Varmus H: Getting ready for gene-based medicine. N Engl J Med 2002; 347:1526-1527Crossref, Medline, Google Scholar
131. Hyman SE: The genetics of mental illness: implications for practice. Bull World Health Organ 2000; 78:455-463Medline, Google Scholar

