
Once-Daily Atomoxetine Treatment for Children and Adolescents With Attention Deficit Hyperactivity Disorder: A Randomized, Placebo-Controlled Study
Abstract
OBJECTIVE: The authors assessed the efficacy of once-daily atomoxetine administration in the treatment of children and adolescents with attention deficit hyperactivity disorder (ADHD). METHOD: In a double-blind study, children and adolescents with ADHD (N=171, age range=6–16 years) were randomly assigned to receive 6 weeks of treatment with either atomoxetine (administered once daily) or placebo. RESULTS: Outcomes among atomoxetine-treated patients were superior to those of the placebo treatment group as assessed by investigator, parent, and teacher ratings. The treatment effect size (0.71) was similar to those observed in previous atomoxetine studies that used twice-daily dosing. Parent diary ratings suggested that drug-specific effects were sustained late in the day. Discontinuations due to adverse events were low (less than 3%) for both treatment groups, and no serious safety concerns were observed. CONCLUSIONS: Once-daily administration of atomoxetine is an effective treatment for children and adolescents with ADHD.
Attention deficit hyperactivity disorder (ADHD) is a common disorder of childhood that affects 3% to 7% of school-age children in the United States. Several pharmacological interventions, including stimulants as well as desipramine and bupropion, have been shown to be effective (1–3). Only stimulants, however, have been approved by the Food and Drug Administration and other regulatory agencies for use in the treatment of ADHD, and there has been strong interest in developing new nonstimulant pharmacologic treatments that would not be controlled substances and that could broaden the therapeutic options available to patients and clinicians.
Atomoxetine, a new investigational compound, has been shown to be efficacious in children and adults with ADHD when administered twice daily (4–6). (Atomoxetine was originally called tomoxetine; the name was recently changed in order to avoid any potential confusion with tamoxifen that might lead to errors in dispensing drug.) Atomoxetine is a potent inhibitor of the presynaptic norepinephrine transporter (Ki 4.5 nM), with minimal affinity for other noradrenergic receptors or for other neurotransmitter transporters or receptors. For most individuals, the plasma half-life of atomoxetine is approximately 4 hours, although approximately 5%–10% of people have a polymorphism at the cytochrome P450 2D6 isoenzyme associated with a longer plasma half-life. As a consequence, the drug does not accumulate in the great majority of individuals when administered twice daily, which was the regimen used in previous studies. Despite this, atomoxetine has been superior to placebo in each of three acute placebo-controlled studies conducted in children and adolescents (4, 5). This observation suggests that factors other than time on receptor could be important determinants of response or that pharmacokinetics in the central nervous system differ from those in the plasma. We hypothesized, therefore, that administering the total daily dose at a single time point could provide satisfactory efficacy for many patients. We report here the results of a randomized, double-blind, placebo-controlled study assessing the efficacy of atomoxetine administered once daily.
Method
This multicenter study was conducted at nine outpatient sites in the United States.
Subjects
Children and adolescents, 6–16 years of age, who met DSM-IV criteria for ADHD, as assessed by clinical interview and confirmed by the Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime Version (K-SADS-PL) (7), were eligible to participate. All patients were required to meet a symptom severity threshold: a score at least 1.5 standard deviations above age and gender norms as assessed by the investigator-administered and -scored parent version of the ADHD Rating Scale–IV (8). Comorbid psychiatric conditions were assessed clinically and with the K-SADS-PL. Important exclusion criteria included serious medical illness, a history of psychosis or bipolar disorder, alcohol or drug abuse within the past 3 months, and ongoing use of psychoactive medications other than the study drug. Patients were recruited by referral and by advertisement.
After description of the procedures and purpose of the study, and before the administration of any study procedure or dispensing of study medication, written informed consent was obtained from each patient’s parent or guardian and written assent was obtained from patients. This study was reviewed by each site’s institutional review board and was conducted in accordance with the ethical standards of the 1975 Declaration of Helsinki, as revised in 2000.
Procedures
All patients underwent a minimum 5-day, medication-free evaluation period, followed by random assignment to either atomoxetine or placebo under double-blind conditions for 6 weeks. Following randomization, patients were seen weekly for two visits and biweekly thereafter. Study drug was administered as a single daily dose in the morning. Patients in the atomoxetine treatment arm began treatment at 0.5 mg/kg/day for 3 days, followed by 0.75 mg/kg/day for the remainder of the first week. The daily dose was then increased to 1.0 mg/kg/day. Four weeks after randomization, patients with a Clinical Global Impression (CGI) severity score >2 (more than minimal symptoms) had a further dose increase to 1.5 mg/kg/day.
Assessments
The protocol-specified primary outcome measure was total score on the ADHD Rating Scale–IV (8, 9), an 18-item scale based on a semistructured interview with the patient’s parent (or primary caretaker) performed by a clinician (nurse, psychologist, social worker, or physician) experienced in working with children with ADHD. Each item corresponds to one of the 18 DSM-IV criteria, and severity for each item is rated on a 4-point scale. Training on the administration of this instrument was conducted for all efficacy raters at the time of the study startup, and all raters were required to rate mock interviews to assess their proficiency before being allowed to rate patients in the study. Other assessments included the Conners’ Parent Rating Scale—Revised: Short Form (10), the Conners’ Teacher Rating Scale—Revised: Short Form (11), and the CGI severity score (12). Additionally, we developed for this study a 13-item parent-rated diary to assess efficacy during evening and early morning periods. The diary was completed daily during the initial medication-free evaluation period as well as during the final visit interval. Each item was rated on a 5-point scale (0=not present, 4=extremely problematic). Symptoms assessed included evening and early morning inattentiveness/distractibility, ability to concentrate on structured tasks, hyperactivity/impulsivity, and oppositionality. Safety and tolerability were assessed by open-ended questioning as well as by regular monitoring of vital signs. Laboratory examinations (including routine chemistries, hematologic measures, and ECGs) were conducted at baseline and endpoint.
Data Analysis
Patient data were analyzed on an intent-to-treat basis. The protocol-specified primary efficacy measure was total score on the ADHD Rating Scale–IV, analyzed with a repeated-measures mixed model that used the MIXED procedure in SAS (13) to compare the treatment groups. The dependent variables were total scores on the ADHD Rating Scale–IV from visit 3 through visit 6 (all postrandomization data). The model also contained fixed-class effect terms for treatment, investigator, visit, and an interaction term between treatment and visit. The model included a random patient effect and total score on the ADHD Rating Scale–IV at baseline (visit 2) as covariates and used an unstructured covariate structure. Protocol-specified definitions of response (25% or greater decrease from baseline in total score on the ADHD Rating Scale–IV) and remission (endpoint CGI severity score of 1 or 2 [no or minimal symptoms]) as well as a post hoc alternative analysis of response, the reliability change index (14), were analyzed by using Fisher’s exact test.
All analyses on secondary measures were performed by using a last-observation-carried-forward approach, and the primary outcome measure (total score on the ADHD Rating Scale–IV) was also analyzed in this fashion. For analysis of last-observation-carried-forward mean change from baseline to endpoint, patients with a baseline and at least one postbaseline measurement were included in the analysis. Treatment comparisons for efficacy measures of mean change from baseline between groups were assessed by using an analysis of variance (ANOVA) model that contained terms for treatment and investigator. For the parent-rated diary, all daily ratings for each item during each interval were averaged and analysis carried out on the mean rating for each item during the visit interval.
To handle missing data for the primary outcome measure, if more than one item of a subscale was missing, then the score for the subscale (and the total score) was also considered missing. However, if a single item was missing, then the mean score for all other items in the subscale was imputed as the score for the missing item when computing the subscale and total scores.
Treatment differences in binary measures between groups were assessed by using Fisher’s exact test. All statistical tests were performed by using a two-sided, 0.05 significance level.
Effect size for the primary outcome measure was calculated as the least square mean treatment difference for atomoxetine versus placebo divided by the square root of the mean square error, based on an ANOVA on last-observation-carried-forward change from baseline scores.
On the basis of previous studies, we calculated that a study group size of 160 patients (80 atomoxetine and 80 placebo) was required. This assumed a detectable effect size of 0.48 on the 18-item investigator-rated ADHD Rating Scale–IV total score; a two-sided, 0.05-level test; and that at most 5% of the enrolled patients would provide no postbaseline information. This study group provided approximately 82% power to detect a treatment difference between atomoxetine and placebo.
Results
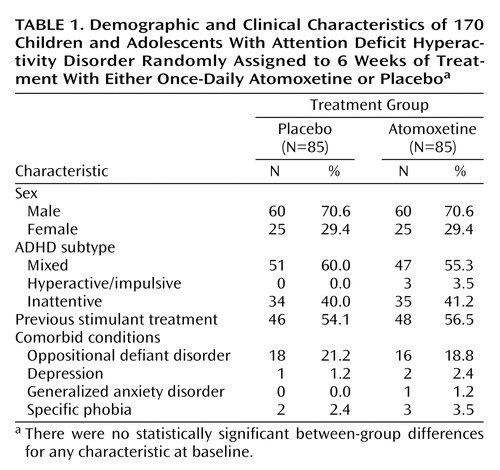
A total of 171 patients were randomly assigned to a treatment group, and 168 were confirmed to have received at least one dose of study drug. One assigned patient did not receive any medication, and it could not be confirmed whether two other assigned patients received study medication. All analyses and summaries excluded the one patient who was confirmed to have not taken study medication, leaving the total reported sample size at 170 (atomoxetine: N=85, placebo: N= 85). The two groups were similar in age (atomoxetine: mean=10.1 years, SD=2.3; placebo: mean=10.5 years, SD=2.5) and other baseline demographic and symptom severity measures (Table 1). The majority of patients included in this study were male. Most patients met criteria for the ADHD mixed or inattentive subtype, with very few patients meeting criteria for the hyperactive/impulsive subtype. Forty-eight patients (56.5%) in the atomoxetine group and 46 (54.1%) in the placebo group reported having been previously treated with a stimulant. The only comorbid diagnosis noted in more than 4% of either group was oppositional defiant disorder.
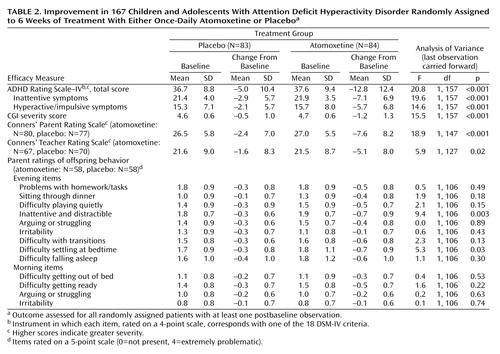
Efficacy results are summarized in Table 2. Mean reductions in the primary outcome measure, total score on the ADHD Rating Scale–IV, were superior for patients randomly assigned to atomoxetine beginning at 1 week and at all subsequent visits (protocol-specified primary analysis [Figure 1]) and were also superior at endpoint according to the protocol-specified secondary analysis (last-observation-carried-forward ANOVA) (Table 2). The treatment effect size was 0.71. At baseline, mean symptom severity (as assessed by ADHD Rating Scale–IV t scores) approached three standard deviations above age and gender norms (atomoxetine group: mean=79.1, SD=12.3; placebo group: mean=78.4, SD=11.7). At endpoint, mean symptom severity was approximately 1.5 standard deviations above age and gender norms for the atomoxetine-treated group (t score change from baseline to endpoint for the atomoxetine group: mean=–13.5, SD=13.7; for the placebo group: mean=–5.4, SD=11.4) (F=18.72, df=1, 157, p<0.001). Response (≥25% reduction from baseline in total score on the ADHD Rating Scale–IV) was superior in the atomoxetine group (59.5%) compared with the placebo group (31.3%) (odds ratio=3.22, 95% CI=1.63–6.41; p≤0.001). Remission (endpoint CGI severity score of 1 or 2) was also superior in the atomoxetine group (28.6%) compared with the placebo group (9.6%) (odds ratio=3.75, 95% CI=1.49–10.30; p=0.003). Response defined by the reliable change index was also superior for atomoxetine (atomoxetine 50%, placebo 25.3% [odds ratio=2.95, 95% CI=1.46–6.01; p=0.001]).
Both attentional and hyperactivity/impulsivity symptom clusters responded to atomoxetine, as assessed by the inattention and hyperactivity/impulsivity subscales of the ADHD Rating Scale–IV. Outcomes were similar in stimulant-naive patients and patients previously treated with stimulants. Superiority to placebo was also demonstrated on the ADHD Index scores of the Conners’ Parent Rating Scale and Conners’ Teacher Rating Scale as well as the CGI severity score. Comparisons of mean change in the individual items of the parent-rated daily diary did not demonstrate any differences between atomoxetine and placebo on early morning behavior but did suggest drug-specific effects on two of the evening items (inattentive symptoms and difficulties settling at bedtime) (Table 2). A post hoc nonparametric comparison (Wilcoxon sign test) found that the number of evening items for which the mean decrease was greater with atomoxetine than with placebo was higher than expected by chance (p<0.04). The mean final dose of atomoxetine was 1.3 mg/kg/day (SD=0.2).
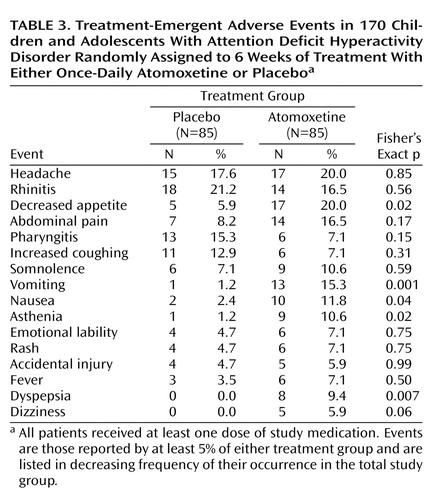
Overall, 73 (86%) of 85 patients randomly assigned to atomoxetine and 75 (87%) of 86 patients assigned to placebo completed the study. Two patients (2%) in the atomoxetine group discontinued because of adverse events (one for vomiting and one for somnolence), as did one patient (1%) in the placebo group (for emotional lability). There were no statistically significant differences in the number of discontinuations between groups overall or for any individual reason. Patients receiving atomoxetine experienced modest mean increases from baseline to endpoint compared with those receiving placebo in both systolic blood pressure (mean change=2.0 mm Hg [SD=8.7] versus –0.7 mm Hg [SD=7.3], respectively; F=4.8, df=1, 165, p<0.04) and pulse (mean change=6.8 bpm [SD=12.0] versus –1.2 bpm [SD=10.3]; F=21.6, df=1, 165, p<0.001). In addition, patients receiving atomoxetine experienced a mean decrease from baseline to endpoint in weight (mean change=–0.9 kg [SD=0.9] versus 0.8 kg [SD=1.1]; F=118.4, df=1, 165, p<0.001). Change in height from baseline to endpoint was similar for the two groups (mean change=0.9 cm [SD=1.3] and 0.8 cm [SD=1.0]; F=0.2, df=1, 150, p<0.65).
Adverse events reported more frequently by the children receiving atomoxetine were primarily gastrointestinal (Table 3), with increases as well in asthenia (this term subsumed reports of tiredness and fatigue). The difference in reports of dizziness approached but did not reach statistical significance. With respect to reports of nausea/vomiting, potentially the most troubling acute effect, most episodes were transient and self-limited (typically 1 to 2 days). Of the 19 patients taking atomoxetine who reported nausea or vomiting, 16 completed the study and chose to continue receiving atomoxetine in an optional open-label extension study.
Discussion
We have previously shown that atomoxetine administered twice daily is an effective treatment for ADHD in children and adults (4, 5, 15). The results of this study are consistent with those findings and extend them by providing evidence of the efficacy and safety of once-daily dosing with atomoxetine, with drug-specific effects persisting late into the day. This is also the first study to provide evidence of the efficacy of atomoxetine based on teacher observations.
Perhaps the most striking finding of this study is the evidence that despite the relatively short plasma half-life of atomoxetine (about 4 hours for most patients), once-daily dosing in the morning was associated with effects that persisted into the evening. This was assessed with a daily diary administered at baseline and during the final visit interval, and the results must be interpreted cautiously, since the instrument is new and its psychometric characteristics unstudied. However, individual item analysis suggests a drug-specific benefit late in the day, with items measuring inattention during late afternoon and early evening and settling at bedtime showing greater improvement for atomoxetine than placebo. Comparisons of mean changes on other individual items and on morning items were not statistically significantly different, but a post hoc nonparametric analysis for the entire group of evening items also suggested an effect in favor of atomoxetine. We note, however, that the clinical importance of this finding and its replicability require further study.
By late afternoon, plasma drug concentrations in most patients are low, and how clinical changes persist after the drug is cleared from the plasma is uncertain. One potential explanation is that the brain kinetics of atomoxetine could differ from those in plasma. Alternatively, atomoxetine’s pharmacologic mechanism (blockade of the presynaptic norepinephrine transporter) may be associated with neuroregulatory changes that last beyond the time the drug is actually on the receptor.
This study did not include a twice-daily dosing arm, and we cannot definitively determine the relative efficacy of once- versus twice-daily atomoxetine administration. However, the primary outcome measure and most study design elements in this study were the same as those of three previous placebo-controlled studies that used twice-daily dosing, and all the investigators in this trial participated in at least one of the previous studies, so preliminary comparisons can be made. In this regard, the treatment effect size in this study for the primary outcome measure was 0.71, compared with 0.77, 0.67, and 0.63 in the studies of atomoxetine given twice daily (4, 5). These data suggest that the magnitude of symptom reduction with once-daily therapy is similar to that of therapy given twice daily, although definitive conclusions will require an adequately sized direct comparison. These results are also comparable to effect sizes reported for methylphenidate on parent-rated measures (16, 17), although any such comparison is limited by differences in study designs and measures, study populations, investigators, etc.
Safety and tolerability were good. Only two children receiving atomoxetine discontinued because of adverse events. Compared with studies in which atomoxetine was given twice daily, there was a slightly greater incidence of pharmacologically expected events, primarily gastrointestinal. These were generally self-limited, and 16 of the 19 children who experienced nausea or vomiting chose to continue taking atomoxetine in an extension study, suggesting that any differences in tolerability between once- and twice-daily administration are of limited clinical importance. Effects on vital signs were similar to those seen in previous studies, with modest increases in cardiovascular tone (pulse and systolic blood pressure) and a mean decrease in weight compared with placebo.
Several factors limit the interpretation of the data presented here. As noted, no direct comparisons of other atomoxetine dosing paradigms or other drugs were included. Also, the dose range used in this study was based on the results of a dose-response study that employed twice-daily dosing, and it is possible that a different range would be optimal for once-daily dosing. Finally, while these results demonstrate efficacy during acute treatment, outcomes during long-term therapy could be different.
These reservations notwithstanding, the results of this study confirm and extend the results of previous studies. Patients often prefer once-daily dosing to more frequent regimens because of its convenience, and the data presented here suggest that for many patients with ADHD, treatment with once-daily atomoxetine will be a viable option.
 |
 |
 |
Received Oct. 3, 2001; revision received May 7, 2002; accepted May 21, 2002. From Lilly Research Laboratories, Indianapolis; Indiana University School of Medicine, Indianapolis; Penn State College of Medicine, Hershey, Pa.; Carolinas Medical Center Department of Psychiatry, Charlotte, N.C.; University of Nebraska Medical Center, Omaha; Mount Sinai School of Medicine, New York; University of Cincinnati Department of Psychiatry, Cincinnati; Clinical Neurophysiology Services PC, Troy, Mich.; Neuroscience Inc., Bethesda, Md.; and Clinical Neuroscience Solutions, Orlando, Fla. Address reprint requests to Dr. Michelson, Lilly Corporate Center, Indianapolis, IN 46285; [email protected] (e-mail). Research was funded by Eli Lilly and Company. The authors thank Denái Milton, Kory J. Schuh, and Michele Y. Hill for their assistance in the preparation of this manuscript.

Figure 1. Improvement in Total Score on the ADHD Rating Scale–IV in Children and Adolescents With Attention Deficit Hyperactivity Disorder Randomly Assigned to 6 Weeks of Treatment With Either Once-Daily Atomoxetine or Placebo
aEach item, rated on a 4-point scale, corresponds with one of the 18 DSM-IV criteria; higher scores indicate greater severity.
bRepeated-measures mixed-model analysis revealed significant between-group difference in mean score reduction (p≤0.001).
1. Wilens TE: The stimulants revisited. Child Adolesc Psychiatr Clin N Am 2000; 9:573-603Crossref, Medline, Google Scholar
2. Biederman J, Baldessarini RJ, Wright V, Knee D, Harmatz JS: A double-blind placebo controlled study of desipramine in the treatment of ADD, I: efficacy. J Am Acad Child Adolesc Psychiatry 1989; 28:777-784Crossref, Medline, Google Scholar
3. Conners CK, Casat CD, Gualtieri CT, Weller E, Reader M, Reiss A, Weller RA, Khayrallah M, Ascher J: Bupropion hydrochloride in attention deficit disorder with hyperactivity. J Am Acad Child Adolesc Psychiatry 1996; 35:1314-1321Crossref, Medline, Google Scholar
4. Heiligenstein J, Spencer T, Faries DE, Biederman J, Kratochvil CJ, Conners CK: Efficacy of atomoxetine vs placebo in pediatric outpatients with ADHD, in Proceedings of the 47th Annual Meeting of the American Academy of Child and Adolescent Psychiatry. Washington, DC, AACAP, 2000Google Scholar
5. Michelson D, Faries DE, Wernicke J, Kelsey DK, Kendrick KL, Sallee FR, Spencer T (Atomoxetine ADHD Study Group): Atomoxetine in the treatment of children and adolescents with ADHD: a randomized, placebo-controlled dose-response study. Pediatrics 2001; 108:e83Google Scholar
6. Spencer T, Biederman J, Wilens T, Prince J, Hatch M, Jones J, Harding M, Faraone SV, Seidman L: Effectiveness and tolerability of tomoxetine in adults with attention deficit hyperactivity disorder. Am J Psychiatry 1998; 155:693-695Link, Google Scholar
7. Kaufman J, Birmaher B, Brent D, Rao U, Ryan N: Kiddie-SADS—Present and Lifetime Version (K-SADS-PL), version 1.0 of October 1996 ed. Pittsburgh, University of Pittsburgh School of Medicine, Department of Psychiatry, 1996Google Scholar
8. DuPaul GJ, Power TJ, Anastopoulos AD, Reid R: ADHD Rating Scale-IV: Checklists, Norms, and Clinical Interpretations. New York, Guilford, 1998Google Scholar
9. Faries DE, Yalcin I, Harder D, Heiligenstein JH: Validation of the ADHD Rating Scale as a clinician administered and scored instrument. J Atten Disord 2001; 5:39-47Crossref, Google Scholar
10. Conners CK, Sitarenios G, Parker JD, Epstein JN: The revised Conners’ Parent Rating Scale (CPRS-R): factor structure, reliability, and criterion validity. J Abnorm Child Psychol 1998; 26:257-268Crossref, Medline, Google Scholar
11. Conners CK, Sitarenios G, Parker JD, Epstein JN: Revision and restandardization of the Conners Teacher Rating Scale (CTRS-R): factor structure, reliability, and criterion validity. J Abnorm Child Psychol 1998; 26:279-291Crossref, Medline, Google Scholar
12. Guy W (ed): ECDEU Assessment Manual for Psychopharmacology: Publication ADM 76-338. Washington, DC, US Department of Health, Education, and Welfare, 1976, pp 218-222Google Scholar
13. SAS/STAT Software: Changes and Enhancements Through Release 6.12, Cary, NC, SAS Institute, 1997Google Scholar
14. Jacobson NS, Truax P: Clinical significance: a statistical approach to defining meaningful change in psychotherapy research. J Consult Clin Psychol 1991; 59:12-19Crossref, Medline, Google Scholar
15. Spencer T, Biederman J, Heiligenstein J, Wilens T, Faries D, Prince J, Faraone SV, Rea J, Witcher J, Zeras S: An open-label, dose-ranging study of atomoxetine in children with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol 2001; 11:251-265Crossref, Medline, Google Scholar
16. Greenhill LL, Swanson JM, Vitiello B, Davies M, Clevenger W, Wu M, Arnold LE, Abikoff HB, Bukstein OG, Conners CK, Elliott GR, Hechtman L, Hinshaw SP, Hoza B, Jensen PS, Kraemer HC, March JS, Newcorn JH, Severe JB, Wells K, Wigal T: Impairment and deportment responses to different methylphenidate doses in children with ADHD: the MTA titration trial. J Am Acad Child Adolesc Psychiatry 2001; 40:180-187Crossref, Medline, Google Scholar
17. Schachter HM, Pham B, King J, Langford S, Moher D: How efficacious and safe is short-acting methylphenidate for the treatment of attention-deficit disorder in children and adolescents? a meta-analysis. Can Med Assoc J 2001; 165:1475-1488Google Scholar

