
Deficient Inhibition as a Marker for Familial ADHD
Abstract
OBJECTIVE: The authors investigated whether deficient inhibitory control, as measured by the stop-signal paradigm, delineates a familial subgroup of attention deficit hyperactivity disorder (ADHD). METHOD: Subjects were 54 ADHD children defined as having poor or good inhibition (on the basis of stop-signal paradigm performance) and 26 healthy comparison children. Family history of ADHD and measures of neurobiological and psychosocial risk were compared among the three groups. RESULTS: ADHD was significantly more prevalent in the families of the children with ADHD who exhibited poor inhibition (48.1%) than in the families of those exhibiting good inhibition (18.5%) or in the families of healthy comparison children (7.7%). No differences in neurobiological or psychosocial risk were found for the three groups. CONCLUSIONS: Deficient inhibition delineates a familial subtype of ADHD. Psychosocial and neurobiological factors did not account for inclusion in the good inhibition group and did not act conjointly with inhibition to increase the risk for ADHD in the poor inhibition group. This study demonstrates that cognitive measures such as a laboratory measure of inhibition can serve as phenotype markers for genetic analyses.
Attention deficit hyperactivity disorder (ADHD) is a prevalent and impairing childhood psychiatric disorder. Genetic factors play a major role in the etiology of the disorder. Findings from family studies indicate a higher incidence of ADHD among first- and second-degree relatives of ADHD individuals (1, 2). Siblings of children with ADHD have two to three times the risk of having ADHD (30%) when compared with siblings of healthy comparison subjects (3). Twin studies indicate that ADHD is a highly heritable condition: the estimated contribution of additive genetic factors is approximately 70% (4). Adoption studies indicate a higher rate of ADHD among biological relatives than among adoptive parents (5).
Molecular research implicates various genetic risk factors, primarily in genes involved in the dopamine system (e.g., dopamine transporter [6] and the dopamine receptor genes D4 [7, 8] and D5[9]). However, results are somewhat variable, and particular gene effects are small, accounting for only a small percent of variance within the disorder (10).
As in other complex, non-Mendelian disorders, a number of factors obscure the role of genetic factors: etiological heterogeneity, reduced penetrance, variable expressivity, and error in defining the behavioral phenotype. It is likely that ADHD is a multigenic and etiologically heterogeneous disorder. Numerous neurobiological and psychosocial risk factors are associated with ADHD, especially those acting during the period of rapid perinatal brain growth (11). These factors include fetal exposure to alcohol, drugs, and tobacco (12, 13) as well as perinatal obstetrical complications (14). Moreover, ADHD could result from an interaction of any combination of environmental or genetic factors, rather than any one particular factor (15).
Concordance for monozygotic twins is substantially less than 100%, which indicates that the genetic influence is not completely penetrant or that the expression of the genetic susceptibility is variable. The genetic susceptibility for ADHD can be expressed in a range of known or currently unknown behavioral and nonbehavioral phenotypes. In addition, genetic risk can be expressed in pathophysiological traits—such as cognitive deficits—rather than, or in addition to, behavioral phenotypes.
Similarly, measurement error in defining the behavioral phenotype can impede identification of the genetic risk factors in ADHD. Varying diagnostic criteria yield markedly different prevalence rates and define groups with different characteristics (16, 17). The ADHD phenotype is further complicated by the delineation of numerous subtypes based on phenomenology (e.g., the inattentive, hyperactive-impulsive, and combined subtypes described in DSM-IV) or comorbid diagnoses (e.g., conduct disorder, reading disabilities). Although these subtypes have distinct clinical correlates, it is not clear whether they share the same genetic risk factors.
These factors can create classes of individuals with the ADHD phenotype but not the genetic susceptibility factors (false positives) or the genetic susceptibility but not the phenotype (false negatives) (18). This makes detection of linkage and localization of susceptibility genes more difficult. These obstacles point to the need for identifying more objective, nonbehavioral indicators that can identify etiologically homogeneous subgroups corresponding to distinct etiological entities (18). These nonbehavioral indicators may point to functional variations of the brain—such as cognitive deficits—or to specific pathology of particular brain regions and neural networks. The goal is to identify indicators that are more closely linked to an underlying genetic susceptibility than the behavioral phenotype (19).
Current formulations of ADHD focus on deficits in various aspects of executive function (20), in particular deficient inhibition (21, 22). Deficient inhibition produces a cascade of secondary impairments in behavior, working memory, self-regulation of affect-motivation-arousal, internalization of speech, and reconstitution. These deficits underlie the behavioral abnormalities exhibited by ADHD individuals in everyday life and the impulsive and inattentive performance exhibited on a variety of laboratory tasks (for review, see reference 23).
Although inhibition has been studied by using a variety of measures, recent investigations of inhibition have relied on the stop-signal paradigm (24). The stop-signal paradigm is a laboratory task that requires a rapid ongoing motor response and the sudden cessation of that response following a specified signal (tone). In one study that used the stop-signal paradigm, a deficit in inhibition among children with ADHD was demonstrated and extensively replicated (25). Neuroimaging studies indicate that the frontostriatal structures implicated in ADHD are activated by the stop-signal paradigm (26). While findings of deficient inhibition in children with ADHD have been robust, not all children with ADHD perform poorly on the task.
The primary aim of the study was to determine if deficient inhibition denotes a familial subgroup of ADHD. If so, we would expect that children with ADHD and deficient inhibition would have a family history in which ADHD was significantly more prevalent than it was in families of children with ADHD who exhibit normal/good inhibition or in families of healthy comparison children. Our secondary hypothesis was that children with ADHD who exhibit good inhibition would have an alternative, nongenetic etiology characterized by greater exposure to environmental or psychosocial risk factors relative to children with ADHD who exhibit deficient inhibition or healthy comparison children.
Method
Subjects
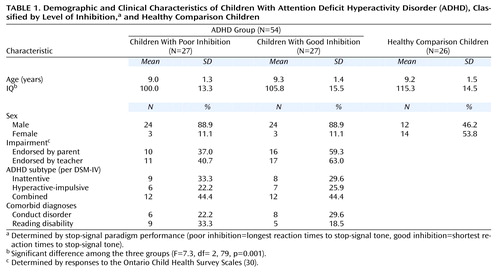
Participants (N=88) were children referred to a child psychiatry clinic in an urban pediatric hospital. Healthy comparison children (N=26) were recruited through advertisements in community newspapers. We excluded participants if they had a history or evidence of neurological disorders such as epilepsy, chronic or serious medical problems, psychosis, clinically significant mood or anxiety disorders, or a verbal or performance IQ of less than 80. Inclusion criterion for the study was age 7 to 12 years, a confirmed diagnosis of ADHD, and valid stop-signal reaction time scores. The healthy comparison children met all the aforementioned criteria with the exception of an ADHD diagnosis; no diagnoses of conduct disorder, reading disabilities, or oppositional defiant disorder were seen in the healthy comparison children. All children were free of psychostimulant medication for 48 hours preceding testing. No children were taking antidepressant medications at the time of the study. Parents gave written consent for the children to participate in the study, and all children gave verbal assent.
Measures
We assigned a diagnosis to each participant on the basis of a semistructured clinical diagnostic interview with the child’s parents (the Parent Interview for Child Symptoms, unpublished 1994 instrument of Schachar and Ickowicz) and with each child’s classroom teacher (Teacher Telephone Interview, unpublished 1990 instrument of Schachar and Tannock). Reliability of the Parent Interview for Child Symptoms is high (kappa=80%) for ADHD. Interviews were administered by experienced psychologists and social workers who were trained to 90% interrater reliability. The Parent Interview for Child Symptoms covers DSM-IV symptoms of ADHD, conduct disorder, oppositional defiant disorder, and anxiety, mood, and other single-symptom and internalizing disorders. The Teacher Telephone Interview covers ADHD, conduct disorder, and oppositional defiant disorder and screens for other disorders. Mothers were the primary informants for diagnostic information. When fathers were present, both parents participated in the interview.
Participants were administered the WISC-III (27) as a measure of intellectual functioning. They were also administered three reading measures: the reading subscale of the Wide-Range Achievement Test 3 (28) and word attack and word identification from the Woodcock Reading Mastery Test—Revised (29).
To be classified as having ADHD, children had to meet DSM-IV criteria for ADHD (defined as at least six of nine symptoms of either inattention or hyperactivity-impulsivity in addition to age at onset and severity criteria). In order to ensure that the pervasiveness criterion of DSM-IV was met, an ADHD diagnosis required presence of ADHD criteria according to interview responses of one informant (either parent or teacher) and a minimum of four inattentive or four hyperactive symptoms according to the other informant.
Clinical Variables
Comorbid diagnoses
Conduct disorder was diagnosed if two or more DSM-IV criteria for conduct disorder were reported by parents or teacher, or if one conduct disorder symptom and full criteria for oppositional defiant disorder were met (four or more symptoms present). Presence of a reading disability was defined by a score 1.5 standard deviations below the mean of any one of the reading measures or by a score of 1 standard deviation below the mean of two of the three reading measures.
Family history
The interviewer asked the parent of each participant about current and past family history of psychiatric, emotional, learning problems, and ADHD. A decision regarding presence or absence of a family history of ADHD was made by the clinician who was blind to diagnostic and inhibitory classification of the child. A family history of ADHD was coded as present if ADHD was identified in the mother, father, or sibling of the child.
Neurobiological and psychosocial risk factors
The Ontario Child Health Survey Scales Family and Household Form (30) was used to record exposure to neurobiological and psychosocial risk factors. Factors were examined individually and then combined into two multivariate risk indexes as recommended by Biederman et al. (31). Each item was given equal weight. Examples of factors included in the neurobiological risk index were perinatal bleeding, high blood pressure, seizures, infection, severe nausea or vomiting, and use of drugs or alcohol. Examples of factors that were included in the psychosocial risk index were living in subsidized housing, single-parent family, overcrowded living arrangements, and separation from parents before the age of 2.
Impairment
Impairment was assessed by using the parent and teacher ratings on the Ontario Child Health Survey Scales. Parents and teachers rated each participant on items concerning the impact of any behavior problems on functioning at home, school, or in the community. Impairment scores were standardized by age and gender by using general population norms.
Measurement of Inhibition
For detailed discussion of the stop-signal paradigm apparatus and stimuli see Schachar et al. (32). Calculation of the stop-signal reaction time uses a tracking algorithm designed to find a stop-signal delay that ties the race between the go process and the stop process. The algorithm increases stop-signal delay if subjects inhibit successfully, and it decreases stop-signal delay if subjects fail to inhibit. If the increments and decrements are equal in magnitude (50-msec changes), the algorithm will converge on a stop-signal delay at which the subjects inhibit 50% of the time. At that point, the go process and the stop process finish at the same time, on average. Go reaction time and stop-signal delay are observable. Stop-signal reaction time is unobservable, but it can be estimated simply by subtracting stop-signal delay from mean go reaction time (33). The stop-signal paradigm shows moderate to high test-retest reliability and stability over repeated administrations (34; unpublished 2001 paper by Tannock).
The experimental groups were derived from the group of 88 children with ADHD. Stop-signal reaction times were divided into thirds to determine groups with extremely long and extremely short stop-signal reaction times. This approach was decided a priori on the basis of the recommendation of Feldt (35) for the examination of extreme groups within a sample. We matched the high and low groups for sex and age. The resulting groups each contained a total of 27 subjects (24 male and three female). Those subjects with stop-signal reaction time scores in the top third of the group (longest stop-signal reaction times) were identified as having poor inhibition. The subjects with stop-signal reaction time scores in the bottom third (shortest stop-signal reaction times) were identified as having good inhibition. The poor inhibition group (mean age=9.0, SD=1.3) had a mean stop-signal reaction time of 510.7 msec (SD=233.2), and the good inhibition group (mean age=9.3, SD=1.5) had a mean stop-signal reaction time of 205.2 msec (SD=35.3). The ADHD subjects in the good inhibition group had a level of inhibitory control similar to that of children of the same age in the general population (36). The mean stop-signal reaction time of the poor inhibition group was 3.8 standard deviations above the mean of the normative sample of the same age group (36). The lowest limit of the poor inhibition group was approximately one standard deviation above the mean stop-signal reaction time found within a normative sample.
The comparison group consisted of 26 children, 12 boys and 14 girls. Within this group, six subjects had stop-signal reaction time scores within the poor inhibition range. This level of poor inhibition is similar to the level reported in a general population sample (36). The mean stop-signal reaction time in the normative sample was 223.0 (SD=75.3), and for the normal comparison group used in the present study it was 257.3 (SD=78.9).
Analysis
Family history of ADHD in each of the groups was compared by using a multinomial regression. Groups were compared by using one-way analysis of variance (ANOVA) to detect differences in neurobiological and psychosocial risk, IQ, and impairment. A likelihood chi-square ratio and one-way ANOVA was used to investigate an association between family history of ADHD and both IQ and gender. In order to determine which variables best predicated inclusion in the poor and good inhibition groups, a logistic regression was used. The effect of comorbidity was assessed informally because of the small size of comorbidity subgroups.
Results
Among the children with ADHD, those with poor and good inhibition did not differ in age, IQ, impairment, ADHD subtype, or comorbid diagnoses (Table 1). An ANOVA indicated significant IQ differences among the three groups (Table 1), with the healthy comparison children having a significantly higher IQ than the children with ADHD who exhibited either poor inhibition (p=0.001, Dunnet’s post hoc test) or good inhibition (p<0.05, Dunnet’s post hoc test). Neither IQ nor gender were associated with family history of ADHD.
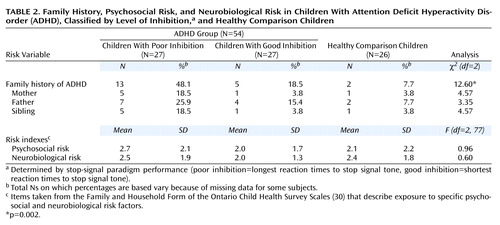
Multinomial logistic regression indicated that the prevalence of ADHD was significantly higher in the families of children with ADHD who exhibited poor inhibition than in the families of children with ADHD who exhibited good inhibition or in the families of the healthy comparison children (Table 2>). Family history of ADHD did not differ between the children with ADHD who exhibited good inhibition and healthy comparison children (Exp[B]=2.72, 95% CI=0.48–15.52, df=1, p<0.26) but did differ between the children with ADHD who exhibited poor inhibition and both those exhibiting good inhibition (Exp[B]=4.09, 95% CI=1.19–13.98, df=1, p<0.03) and the healthy comparison children (Exp[B]=11.14, 95% CI=2.19–56.76, df=1, p=0.004). As seen in Table 2, similar (although statistically nonsignificant) differences in ADHD prevalence rates were seen in the three groups when family history was examined specifically for mothers (p<0.11), fathers (p<0.19), and siblings (p<0.11).
No significant differences were found between the three groups on the neurobiological and psychosocial risk indexes (Table 2). Analysis of individual neurobiological risk factors indicated that two of 21 risk factors were significantly more common in the ADHD subjects with poor inhibition: maternal emotional stress during pregnancy (χ2=7.29, df=1, p=0.007) and jaundice (χ2=4.48, df=1, p<0.04). One of 21 psychosocial risk factors (mother limited in daily activities because of a medical condition) was found to be significantly more common in the ADHD subjects with poor inhibition (χ2=5.55, df=1, p<0.02). This number of significant results (one or two out of 21) would be expected by chance.
The logistic regression involving family history of ADHD and neurobiological and psychosocial risk indexes indicated that the only variable that significantly distinguished membership in the poor and good inhibition groups was a family history for ADHD (Exp[B]=4.09, 95% CI=1.20–14.00, df=1, p=0.05).
In order to assess the specificity of the association of stop-signal reaction time and family history, the go response times of the children with ADHD were divided as was done for the stop-signal reaction times. Family history of ADHD did not differ between subjects with long and short go response times (33.3% and 34.6%, respectively).
In order to determine any possible association of comorbidity with family history of ADHD, family history was investigated in subsets of subjects from both the poor and good inhibition groups with either comorbid conduct disorder or a reading disability. Because of the small size of the comorbidity subgroups, the comorbidity data was investigated informally. The distinction of poor versus good inhibition predicted ADHD prevalence in the families of subjects with comorbid conduct disorder (50.0% and 25.0%, respectively), a comorbid reading disability (44.4% and 20.0%), and pure (i.e., no conduct disorder or reading disability) ADHD (50.0% and 14.3%) at rates comparable to that of the entire cohort (48.1% and 18.5%).
Discussion
Using family history as a proxy for genetic risk (14, 18), we found that a deficit in inhibitory control identified a subgroup of children with ADHD that had greater risk for familial ADHD. Children with ADHD who exhibited poor inhibition were significantly more likely to have a first-degree relative with ADHD than were children with ADHD who exhibited good inhibition.
The association between poor inhibition and family history of ADHD was not the result of differences between groups in impairment, severity or type of ADHD symptoms, age, sex, or IQ. Similarly, the difference between those with good and poor inhibition was not an artifact of comorbidity. Although comorbidity was investigated informally, the results indicated that either with or without a comorbid diagnosis (conduct disorder or a reading disability), those with poor inhibition had an equally high familial risk of ADHD. The speed of responding (go response) was investigated, and no association was found, indicating that findings were specific to inhibition rather than an artifact of generalized slowing of all responses in the ADHD children.
The apparent similarity in behavior, comorbidity, age, and gender of those with good and poor inhibition suggests that ADHD is a final common pathway of various etiological mechanisms. These various pathways result in behavioral syndromes that are similar insofar as we could discern. Findings from this study indicated that the etiology of one of these pathways involves deficient inhibition, and, presumably, this pathway could be mediated by genetic susceptibility factors as evident in the greater familial risk for ADHD.
The specific mechanism by which these genes influence the development of ADHD is unknown. Several genetic risk factors within the pathway of catecholamine synthesis have been suggested from molecular genetic studies (i.e., dopamine transporter 1, D4 dopamine receptor gene). Differential function of proteins involved in these pathways as a result of different polymorphisms or mutations in the respective genes could be responsible for the phenotype. For example, less sensitive dopamine receptors might be expressed in basal ganglia and frontal regions involved in executive functions. According to this logic, genetic susceptibility factors may be expressed in specific indicators of brain function such as deficient inhibition.
Genetic factors are presumably not the only factors that can result in abnormalities of the neural substrate of inhibition. Acquired neurobiological or psychosocial risk factors could cause the same or similar abnormality (37). If so, we might have expected a higher rate of these factors in children with poor inhibition than in those with good inhibition. That was not found among the factors examined in this study. This absence of an association between poor inhibition and acquired neurobiological risk is not surprising, since clear evidence of traumatic brain injury is a typical exclusion criteria for ADHD studies, as it was in this study. Alternatively, we might have expected that children with poor inhibition who did not have a family history of ADHD would be at a greater risk for neurobiological or psychosocial adversity. This was not the case. Another explanation for the more than 50% of children with poor inhibition not having a family history of ADHD might be that the positive and negative classifications for family history of ADHD were erroneous. Finally, the measure of inhibition may itself have been inaccurate to some extent. We used only a single measure of inhibitory control, and the threshold that was set to distinguish subjects with good and poor inhibition (top and bottom third of reaction times) may not have been an optimal approach. Other measures may increase demand for inhibitory control and identify more individuals with an inhibition deficit and, as a consequence, more individuals with a positive family history. Despite all these sources of potential error, the finding was quite clear. Half of the children with ADHD who exhibited poor inhibition had a positive family history of ADHD, a prevalence that is much higher than seen within the ADHD population.
Although family history can be positive as a result of psychosocial factors operating within the environment, the strong evidence of genetic effects in ADHD and the relative absence of environmental effects (38) lend support to a genetic mode of familial transmission. Indeed, the good and poor inhibition groups did not differ in the level of psychosocial or neurobiological risk factors, indicating that it is unlikely the greater risk of a positive family history observed among those exhibiting poor inhibition was a result of these types of risks.
If poor inhibition indicates a genetic risk for ADHD, what accounts for the disorder in the good inhibition group? First, it must be pointed out that the children with ADHD who exhibited good inhibition were more likely than were the healthy comparison children to have a positive family history of ADHD, although the rate was substantially less than that found among the children with ADHD who exhibited poor inhibition. The children with good inhibition may have been affected by genetic susceptibility factors that are less penetrant than those that affected the children with poor inhibition. Second, there may be nongenetic factors that cause a phenocopy of ADHD through alternative pathways or through an interaction with genetic susceptibility. We found no support in the factors we examined for an alternative pathway among the children with ADHD who exhibited good inhibition. Exposure to these risks was reported equally in all three groups.
The primary limitation of this study was the use of a family history method, which is highly dependent on the number of individuals in a proband’s family and the reliability of the informant. Also, the small size of the comorbidity subgroups did not allow for formal analysis of the effect of conduct disorder or a reading disability on family history. The findings of this study are preliminary and replication is warranted. Future research would benefit from including measures of inhibitory control on family members of ADHD probands as well as comparison subjects and their families. Unaffected family members may actually have poor inhibition and would be more correctly considered affected. Further investigation is warranted in children with ADHD who exhibit good inhibition to determine what factors contribute to the development of the disorder in those individuals.
Conclusions
This study demonstrates that cognitive measures such as a laboratory measure of inhibition can serve as phenotype markers for genetic analyses. The long-term goal of this approach is to develop nonbehavioral indicators that can increase the power of the search for genetic susceptibility factors in ADHD. These indicators can be used to refine criteria for affected status of probands and siblings through the inclusion of deficient inhibition as a criterion, or as a quantitative trait phenotype, in place of or in combination with the ADHD phenotype.
 |
 |
Received Aug. 4, 2000; revision received April 11, 2001; accepted May 10, 2001. From the Department of Psychiatry and the Brain and Behavior Program at The Hospital for Sick Children; and the Ontario Institute for Studies in Education, University of Toronto, Toronto. Address reprint requests to Dr. Schachar, Department of Child Psychiatry, The Hospital for Sick Children, 555 University Ave., Toronto, ONT M5G 1X8, Canada; [email protected] (e-mail). Supported by a grant from the National Health Research Development Program of Health Canada (6606-5612-401). The authors thank George Whiting, Rosemary Tannock, Gordon Logan, and Cathy Barr for their contribution to this research.
1. Faraone SV, Biederman J, Chen WJ, Milberger S, Warburton R, Tsuang MT: Genetic heterogeneity in attention-deficit hyperactivity disorder (ADHD): gender, psychiatric comorbidity, and maternal ADHD. J Abnorm Psychol 1995; 104:334-345Crossref, Medline, Google Scholar
2. Hudziak J, Todd RD: Familial subtyping of ADHD. Current Opinion in Psychiatry 1993; 6:489-493Crossref, Google Scholar
3. Biederman J, Faraone SV, Keenan K, Benjamin J, Krifcher B, Moore C, Sprich-Buckminster S, Ugaglia K, Jellinek MS, Steingard R, Spencer T, Norman D, Kolodny R, Kraus I, Perrin J, Keller MB, Tsuang MT: Further evidence for family-genetic risk factors in attention deficit hyperactivity disorder: patterns of comorbidity in probands and relatives in psychiatrically and pediatrically referred samples. Arch Gen Psychiatry 1992; 49:728-738Crossref, Medline, Google Scholar
4. Gjone H, Stevenson J, Sundet JM: Genetic influence on parent-reported attention-related problems in a Norwegian general population twin sample. J Am Acad Child Adolesc Psychiatry 1996; 35:588-596Crossref, Medline, Google Scholar
5. van den Oord EJ, Boomsma DI, Verhulst FC: A study of problem behaviors in 10- to 15-year-old biologically related and unrelated international adoptees. Behav Genet 1994; 24:193-205Crossref, Medline, Google Scholar
6. Cook EH Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kieffer JE, Leventhal BL: Association of attention-deficit disorder and the dopamine transporter gene. Am J Hum Genet 1995; 56:993-998Medline, Google Scholar
7. Barr CL, Wigg KG, Bloom S, Schachar R, Tannock R, Roberts W, Malone M, Kennedy JL: Further evidence from haplotype analysis for linkage of the dopamine D4 receptor gene and attention-deficit hyperactivity disorder. Am J Med Genet 2000; 96:262-267Crossref, Medline, Google Scholar
8. Smalley SL, Bailey JN, Palmer CG, Cantwell DP, McGough JJ, Del’Homme MA, Asarnow JR, Woodward JA, Ramsey C, Nelson SF: Evidence that the dopamine D4 receptor is a susceptibility gene in attention deficit hyperactivity disorder. Mol Psychiatry 1998; 3:427-430; correction, 1999; 4:100Google Scholar
9. Barr CL, Wigg KG, Feng Y, Zai G, Malone M, Roberts W, Schachar R, Tannock R, Kennedy JL: Attention-deficit hyperactivity disorder and the gene for the dopamine D5 receptor. Mol Psychiatry 2000; 5:548-551Crossref, Medline, Google Scholar
10. Castellanos FX, Lau E, Tayebi N, Lee P, Long RE, Giedd JN, Sharp W, Marsh WL, Walter JM, Hamburger SD, Ginns E, Rapoport JL, Sidransky E: Lack of an association between a dopamine-4 receptor polymorphism and attention-deficit/hyperactivity disorder: genetic and brain morphometric analyses. Mol Psychiatry 1998; 3:431-434Crossref, Medline, Google Scholar
11. Lou HC: Etiology and pathogenesis of attention-deficit hyperactivity disorder (ADHD): significance of prematurity and perinatal hypoxic-haemodynamic encephalopathy. Acta Paediatr 1996; 85:1266-1271Crossref, Medline, Google Scholar
12. Aronson M, Hagberg B, Gillberg C: Attention deficits and autistic spectrum problems in children exposed to alcohol during gestation: a follow-up study. Dev Med Child Neurol 1997; 39:583-587Crossref, Medline, Google Scholar
13. Schneider ML, Roughton EC, Lubach GR: Moderate alcohol consumption and psychological stress during pregnancy induce attention and neuromotor impairments in primate infants. Child Dev 1997; 68:747-759Crossref, Google Scholar
14. Milberger S, Biederman J, Faraone SV, Guite J, Tsuang MT: Pregnancy, delivery and infancy complications and attention deficit hyperactivity disorder: issues of gene-environment interaction. Biol Psychiatry 1997; 41:65-75Crossref, Medline, Google Scholar
15. Kinney DK, Levy DL, Yurgelun-Todd DA, Tramer SJ, Holzman PS: Inverse relationship of perinatal complications and eye tracking dysfunction in relatives of patients with schizophrenia: evidence for a two-factor model. Am J Psychiatry 1998; 155:976-978Link, Google Scholar
16. Schachar R, Logan GD: Impulsivity and inhibitory control in normal development and childhood psychopathology. Dev Psychol 1990; 26:710-720Crossref, Google Scholar
17. Taylor E: Clinical foundations of hyperactivity research. Behav Brain Res 1998; 94:11-24Crossref, Medline, Google Scholar
18. Tsuang MT, Faraone SV, Lyons MJ: Identification of the phenotype in psychiatric genetics. Euro Arch Psychiatry Clin Neurosci 1993; 243:131-142Crossref, Medline, Google Scholar
19. Matthysse S: Genetic linkage and complex diseases. Genet Epidemiol 1990; 7:29-31Crossref, Google Scholar
20. Denckla MB: Research on executive function in a neurodevelopmental context: application of clinical measures. Dev Neuropsychol 1996; 12:5-15Crossref, Google Scholar
21. Barkley RA, Biederman J: Toward a broader definition of the age-of-onset criterion for attention-deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 1997; 36:1204-1210Crossref, Medline, Google Scholar
22. Quay HC: Inhibition and attention deficit hyperactivity disorder. J Abnorm Child Psychol 1997; 25:7-13Crossref, Medline, Google Scholar
23. Pennington BF, Ozonoff S: Executive functions and developmental psychopathology. J Child Psychol Psychiatry 1996; 37:51-87Crossref, Medline, Google Scholar
24. Logan GD, Kantowitz BH, Riegler GL: On the Ability to Inhibit Selectively: Global and Local Mechanisms of Response Interdiction. Champaign, University of Illinois, 1985, pp 1-37Google Scholar
25. Oosterlaan J, Sergeant JA: Inhibition in ADHD, aggressive, and anxious children: a biologically based model of child psychopathology. J Abnorm Child Psychol 1996; 24:19-36Crossref, Medline, Google Scholar
26. Rubia K, Overmeyer S, Taylor E, Brammer M, Williams SC, Simmons A, Bullmore E: Hypofrontality in attention deficit hyperactivity disorder during higher-order motor control: a study with functional MRI. Am J Psychiatry 1999; 156:891-896Link, Google Scholar
27. Wechsler D: Wechsler Intelligence Scale for Children, 3rd ed. San Antonio, Tex, Harcourt Brace, 1991Google Scholar
28. Wilkinson GS: Wide-Range Achievement Test 3: Administration Manual. Wilmington, Del, Wide Range, 1993Google Scholar
29. Woodcock RW: Woodcock Reading Mastery Test—Revised. Circle Pines, Minn, American Guidance Service, 1987Google Scholar
30. Boyle MH, Offord DR, Racine Y, Szatmari P, Fleming JE, Sanford M: Identifying thresholds for classifying childhood psychiatric disorder: issues and prospects. J Am Acad Child Adolesc Psychiatry 1996; 35:1440-1448Crossref, Medline, Google Scholar
31. Biederman J, Milberger S, Faraone SV, Kiely K, Guite J, Mick E, Ablon S, Warburton R, Reed E: Family-environment risk factors for attention-deficit hyperactivity disorder: a test of Rutter’s indicators of adversity. Arch Gen Psychiatry 1995; 52:464-470Crossref, Medline, Google Scholar
32. Schachar R, Mota VL, Logan GD, Tannock R, Klim P: Confirmation of an inhibitory control deficit in attention-deficit/hyperactivity disorder. J Abnorm Child Psychol 2000; 28:227-235Crossref, Medline, Google Scholar
33. Logan GD: On the ability to inhibit thought and action: a users’ guide to the stop signal paradigm, in Inhibitory Processes in Attention, Memory, and Language. Edited by Dagenbach D, Carr TH. San Diego, Academic Press, 1994, pp 189-239Google Scholar
34. Kindlon D, Mezzacappa E, Earls F: Psychometric properties of impulsivity measures: temporal stability, validity and factor structure. J Child Psychol Psychiatry 1995; 36:645-661Crossref, Medline, Google Scholar
35. Feldt LS: The use of extreme groups to test for the presence of a relationship, in Research Problems in Psychology. Edited by Badia P, Haber A, Runyon R. Reading, Mass, Addison-Wesley, 1970, pp 133-143Google Scholar
36. Williams BR, Ponesse JS, Schachar RJ, Logan GD, Tannock R: Development of inhibitory control across the life span. Dev Psychol 1999; 35:205-213Crossref, Medline, Google Scholar
37. Gerring JP, Brady KD, Chen A, Vasa R, Grados M, Bandeen-Roche KJ, Bryan RN, Denckla MB: Premorbid prevalence of ADHD and development of secondary ADHD after closed head injury. J Am Acad Child Adolesc Psychiatry 1998; 37:647-654Crossref, Medline, Google Scholar
38. Eaves LJ, Silberg JL, Meyer JM, Maes HH, Simonoff E, Pickles A, Rutter M, Neale MC, Reynolds CA, Erikson MT, Heath AC, Loeber R, Truett KR, Hewitt JK: Genetics and developmental psychopathology, 2: the main effects of genes and environment on behavioral problems in the Virginia Twin Study of Adolescent Behavioral Development. J Child Psychol Psychiatry 1997; 38:965-980Crossref, Medline, Google Scholar

