
How and Why Genetic Linkage Has Not Solved the Problem of Psychosis: Review and Hypothesis
Abstract
Objective: The author examined the chromosomal linkage method as an approach to the genetic basis of schizophrenia and bipolar disorder. Method: Comparisons were conducted of recent meta-analyses of genome scans of schizophrenia and bipolar disorder and of the three largest (N>300) sibling pair studies of schizophrenia and schizoaffective disorder and a comparable study of bipolar illness. Results: Recent meta-analyses have not identified consistent sites of linkage. The three largest studies of schizophrenia fail to agree on a single locus, no commonality with bipolar illness has been demonstrated, and there is no replicable support for any of the current candidate genes. Discussion: An alternative to the concept that DNA sequence variation lies in “multiple genes of small effect” is the hypothesis that the variation is epigenetic but related to the genetic transition (“the speciation event”) that separated Homo sapiens from a prior hominid species. This hypothesis draws attention to the chromosomal rearrangement (the Xq21.3/Yp translocation) that occurred some 6 million years ago in the hominid lineage and subsequent rearrangements, including a paracentric inversion, that have taken place within the translocated segment. Here it is argued that the most recent of these events is relevant to specifically human characteristics, including language. The gene pair protocadherin X and Y within this region is under new selective pressure and is in a novel (epigenetic) situation with respect to X inactivation. Conclusions: Epigenetic variation associated with chromosomal rearrangements that occurred in the hominid lineage and that relates to the evolution of language could account for predisposition to schizophrenia and schizoaffective and bipolar disorder and failure to detect such variation by standard linkage approaches.
The search for linkage is the attempt to locate predisposing genes by identifying sites (polymorphisms) in the genome at which sequence variations (alleles) travel with illness within families that have more than one affected member. It is thus a method of locating a disease-inducing gene at a specific site on a particular chromosome.
It has been nearly two decades since Sherrington et al. (1) reported in Nature that they had identified a susceptibility locus for schizophrenia on the long arm of chromosome 5. In seven families that included 39 individuals with DSM-III diagnoses of schizophrenia or schizophreniform disorder, these authors recorded lod scores ranging from 3.22 to 6.49 with different analyses and inclusion or exclusion of cases. They concluded that their findings provided the “first strong evidence for the involvement of a single gene in the causation of schizophrenia.” Within 2 years, however, a number of tests of linkage in the same region (e.g., references 2–5) had been conducted without yielding comparable findings.
Egeland et al. (6) , studying affective disorders, reported that “an analysis of the segregation of restriction fragment length polymorphisms in an Old Order Amish pedigree has made it possible to localize a dominant gene conferring a strong predisposition to manic depressive disease to the tip of the short arm of chromosome 11”—but then reported 2 years later (7) that a reanalysis that included several new individuals and two changes in clinical status “markedly reduces the probability of linkage” and that, indeed, linkage could be excluded from that region. Shortly afterward, the same journal published a paper (8) linking bipolar affective illness to color blindness and glucose-6-phosphate dehydrogenase deficiency as markers on the long arm of the X chromosome with lod scores between 7.52 and 9.17—“that is, the odds in favor of linkage range between 3×10 7 and 10 9 to 1.” No magnitude of lod score comparable to those reported in these early studies is now seriously entertained. Nevertheless, the case for a genetic contribution to schizophrenia (9) and manic-depressive illness is strong (10) .
The question in the light of the subsequent literature is how such apparently convincing evidence could have been obtained. Given this history and the accumulation of much data from studies with larger sample sizes, it is surprising that a claim for linkage with a lod score of 6.5 of schizophrenia to a locus on chromosome 1q21-q22 in a study of 22 families appeared in Science in April 2000 without any clear explanation of the discrepancy with the more modest findings that had by then accumulated (11) . The subsequent failure to replicate the finding in a large collaborative study (12) could perhaps have been predicted.
Method and Results
Meta-Analyses of Genome Scans
Scientific progress requires replication. What has made replication an attainable target in linkage studies has been the pursuit of systematic genome scans in which spaced markers throughout the genome—a hypothesis-free approach—are evaluated against the transmission of disease within families, generally including affected pairs of siblings. In principle, such studies have the potential to reveal linkage to multiple genes, and with increasing sample size, they can be expected to show increasing uniformity of findings across studies. There have now been two meta-analyses of the genome scans in relation to schizophrenia and two in relation to bipolar disorder ( Table 1 ).

Taken together, the findings are instructive:
1. One meta-analysis of schizophrenia draws attention to three and the other to nine possible loci of significance. Badner and Gershon (13) pointed to 8p, 13q, and 22q (p and q indicating the short and long arms, respectively, of the relevant chromosomes) as the regions of greatest significance, whereas Lewis et al. (14) , by the method they adopted (16) , concluded that their strongest finding was on 2p, with significant evidence for linkage on 5q, 3p, 11q, 6p telomere, chromosome 1 at the centromere, and 22 at the short arm telomere all yielding stronger evidence than the strongest candidate, 8p, in the first study. Thus the two meta-analyses applied to a material with an overlap of approximately 500 pedigrees out of a total of 681 (13) or 1,208 (14) agree on only two out of 10 chromosome arms as candidates to harbor a locus for schizophrenia. The strongest finding in one study (14) (linkage to 2p) was not detected in the other analysis; this locus emerged only from a single large study (17) (382 sib pairs) that was included in the second but not the first meta-analysis.
2. While the second meta-analyses of schizophrenia (14) and of bipolar disorder (15) were conducted with an identical methodology, the conclusions that the authors drew are quite different. Whereas the schizophrenia meta-analysis concluded that there were eight bins that yielded genomewide evidence of linkage, the bipolar meta-analysis revealed no such evidence (the locations listed in Table 1 for this study are the nominal 5% by-bin significances), and the locations that yielded the largest rank sums showed no overlap with the bins identified for schizophrenia. This is surprising in light of evidence that there is some genetic commonality between these major phenotypes (18 – 21) , and all the more surprising given that the core phenotype in meta-analysis included diagnoses of schizoaffective disorder.
3. The two findings in the first analysis of bipolar disorder (13) of 353 families were not replicated in the second meta-analysis (15) , which included 347 families from essentially the same published literature but identified four different locations of interest, although none reached genomewide significance by strict criteria.
Comparison of Four Large Linkage Studies
There have now been claims for linkage of schizophrenia to locations on 21 of the 23 pairs of chromosomes, but for any one of these claims the majority of genome scans has failed to detect linkage. Figure 1 presents a comparison of the findings of DeLisi et al. (17) , Williams et al. (22) , and Suarez et al. (23) , the three largest linkage studies published thus far that include schizophrenic and schizoaffective psychosis, together with findings from a study of bipolar illness of comparable size and design (Lambert et al. [24] ). The first two studies (17 , 22) each draw attention to two loci—the first to loci on chromosomes 2 and 10, and the second to loci on chromosomes 10 and 17, with the loci on chromosome 10 being located at the short arm and long arm telomeres, respectively—and the third (23) points to loci on chromosomes 5, 8, 10, and 11. The findings in each study are weak (lod scores of less than 3 are generally taken as lacking significance), and there is no agreement on a single locus between the three studies of schizophrenia and schizoaffective psychosis and no commonality with the findings in bipolar disorder (modest peaks on chromosomes 4 and 6). Moreover, none of the studies gives support to a number of candidate genes that have recently been suggested (25) , such as RGS4 and DISC1 (on chromosome 1), dysbindin (on chromosome 6), neuregulin (on chromosome 8), DAAO (on chromosome 12), G72 (on chromosome 13), and COMT (on chromosome 22). If any of these candidates (identified on the basis of other linkage studies) were really associated with disease, why would this association not be apparent in these four largest studies?

a DeLisi et al. (17), Williams et al. (22), Suarez et al. (23) (each including cases of schizophrenia and schizoaffective disorder), and Lambert et al. (24) (bipolar disorder and schizoaffective disorder) are compared. Maximum multipoint lod scores are plotted for each chromosome as a function of distance in centimorgans (cM) from the short arm telomere. Also shown are the consensus peaks identified in the meta-analyses of Badner and Gershon (13) (downward arrows) and Lewis et al. (14) (upward arrows—the six loci indicated relate to the six bins reported as yielding genomewide evidence for linkage) and the “candidate genes” of Harrison and Owen (26) (diamonds), such as RGS4 and DISC1 on chromosome 1, dysbindin on chromosome 6, neuregulin on chromosome 8, DAAO on chromosome 12, G72 on chromosome 13, and COMT on chromosome 22.
The inconsistency of the findings in these comparisons deserves emphasis. First, there is no agreement between these three large linkage studies, each conducted with broadly similar techniques on samples of sibling pairs identified by comparable diagnostic criteria. Second, there is no consistency in the findings of two meta-analyses conducted on a body of literature that included a core of the same studies. Third, there is no relationship between the locations identified in either of the meta-analyses and the modest peaks that emerged from the three sibling pair studies summarized here. The eye is drawn to chromosome 8 as a possible exception, but the peak in the study of Suarez et al. (23) has no counterpart in the other two linkage studies. Fourth, the candidate genes that have attracted widespread interest in the recent literature are not in any systematic way related either to the locations identified in the meta-analyses or to any conjunction of peaks from the three largest sibling pair studies. Again in relation to chromosome 8, Suarez et al. (23) conclude that the broad peak that emerged from their studies was clearly telomeric to neuregulin, and the same authors (Duan et al. [26] ) have conducted a systematic association study of this region with negative outcome.
Nor do studies of genetic isolates clarify the situation. There have now been nine claims for linkage in such populations (see Table 3 in DeLisi et al. [27] ), but in general there is no agreement between isolates and no obvious agreement between any one of these claims and the consensus of either of the meta-analyses.
Discussion
Where does this leave the search for genes for the spectrum or continuum of psychosis? At the very least, it is clear that no strong signal is emerging, and more important, there is no obvious criterion for distinguishing signal from noise. One possible conclusion is that many genes of small effect are relevant, that there is significant heterogeneity across populations, and that samples much larger than those currently available will be required to detect reliable linkage. While this view (“multiple genes of small effect”) is popular among psychiatric geneticists, there is no indication at all in the current literature that it has a chance of success. The field of psychosis genetics, as based on linkage studies, is thus at an impasse—there is no clear way ahead.
A quite different answer is conceivable: that the genetic variation is relatively homogeneous in form but of a nature that is in principle invisible to the linkage approach. It is possible that the relevant variation is epigenetic, that is, that it lies in modifications, such as methylation of the sequence or in variations in methylation, phosphorylation, and acetylation of the associated histones that are components of the chromosome, rather than in alterations of the DNA sequence itself, and for this reason is not detected by the linkage strategy.
There are already indications of this in the data from monozygotic twins, the cornerstone of the evidence for the genetic theory. Whereas concordance (40%–50%) is greater than that seen in dizygotic twins (12%–15%), consistent with a genetic contribution, it falls well short of 100%. The discrepancy is often interpreted as evidence for an environmental interaction, but no consistent differences in exposure to putative risk factors between affected and unaffected members of discordant pairs have been identified (28) . The alternative is that discordance reflects stochastic differences in gene expression in the course of development. The steep decline from around 10% in first-degree relatives to between 1% and 2% in second-degree relatives, close to the risk in the general population, is often argued to be consistent with “multiple genes of small effect” (29) but is also compatible with a genetic imprint imposed or reimposed at meiosis with a short half-life between generations.
The Case for a Single Gene
Contrary to recent interpretations of linkage findings, there are arguments for a single gene underlying psychotic disorder (e.g., reference 30 ):
1. The structural changes in the brain in psychotic illness are relatively homogeneous. There is a degree of ventricular enlargement (31) , and the range of values in the patient group reflects an upward shift of the distribution relative to controls, without an increase in variance (32) . Ventricular enlargement is thus a consistent correlate of psychosis. It has been demonstrated in a number of ethnically separated groups. It is unclear how this could be the result of a number of unrelated genes varying independently in different populations.
2. Incidences are relatively uniform across populations, particularly when the core (nuclear) symptoms are assessed (33) . This is true also when bipolar illness is the case definition (34) . The fact that incidence is apparently unrelated to geographic or other environmental variation (see references 30 and 35 for a discussion of the absence of evidence for environmental factors) is the key observation that has to be explained by theories of etiology. If psychosis is due to multiple genes, why should these not vary independently in separated populations?
3. Although it has been claimed in the past that certain forms of psychosis (e.g., periodic catatonia and cycloid psychosis) “breed true” within families, this claim is not defended in the contemporary literature. What is observed is that the form of psychosis varies within families encompassing individuals with schizoaffective and affective illness as well as more characteristically nuclear forms of schizophrenia. Each of the larger collaborative studies of both schizophrenic and affective psychoses includes schizoaffective illnesses in the core phenotype (see also Table 1 ). This suggests some sort of a continuum. If different genes were responsible for different psychotic illnesses in different families, a homogeneity of the clinical picture within families would be expected, and this is not observed.
4. In a graphic analysis of the relationship between penetrance and concordance in monozygotic and dizygotic twins, Pritchard (36) has shown that only a single-gene model is consistent with the data. Polygenic models predict greater discrepancies between monozygotic and dizygotic concordance than are observed.
5. It is necessary to postulate a significant genetic advantage to account for the persistence of psychosis in association with a fecundity disadvantage. If two genes were relevant, it is unlikely that the balance of advantage and disadvantage associated with each would be similar (37) . One gene would be selected out.
Any argument for a single gene must explain why such a gene has not long since been detected by the linkage approach. The answer proposed here is that the variation relates to the characteristic that separates Homo sapiens as a species from other primate and hominid species, and that interindividual variation is epigenetic in form. The symptoms of psychosis can readily be seen as disturbances of the H. sapiens -specific capacity for language. Hallucinations (voices), disturbances of thought processes (thoughts experienced as alien, loss of direction), and even delusions (distortions of meaning) can all be conceived as deviations in the transition from thought to speech production or from perceived speech to meaning (38 , 39) . Thus the phenomena of psychosis are associated with the core characteristic that defines the species. The function to be accounted for is language, and this faculty has long posed difficulties for evolutionary theory. It represents an apparent discontinuity or saltation that is not easily assimilated by the conventional gradualist view that species separate in response to differential environmental selective pressures. That the variation associated with psychosis relates precisely to those changes that distinguish H. sapiens from prior hominid species raises the question of the identity of the species transition.
What Is Characteristic of the Human Brain?
According to the Out of Africa hypothesis (40) , all modern human populations have an origin in Africa sometime between 100,000 and 150,000 years ago, at which time a species arose that replaced prior hominid species such as H. Heidelbergensis , H. erectus , and the Neanderthals ( Figure 2 ). This species had the capacity to represent the environment in symbols, a capacity manifest in language that can be traced back in the archaeological record through artifacts to 90,000 years ago, but not much earlier (41 , 42) . It is parsimonious to assume that the transition to language and the origin of the species reflect the same genetic event. To judge by the relative stability of the pattern of artifacts associated with preceding hominid species, the transition was relatively abrupt.

a The hypothesis is that current human populations arose as a result of a genetic transition that occurred in Africa between 100,000 and 150,000 years ago and that modern Homo sapiens replaced hominid species such as Homo erectus and the Neanderthals that had already moved out of Africa. It is plausible that the genetic change (the “speciation event”) that facilitated the transition was responsible for the modern human capacity for language and symbolic representation. (For arguments concerning the timing and the abruptness of the transition, see references 41, 42, and 73; for a discussion of preceding hominid species, see reference 74.)
What can this genetic event have been? Broca (43) , following Dax (44) , reported that language production in the frontal lobes was located on the left side. In 1877 he wrote, “Man is, of all the animals, the one whose brain in the normal state is the most asymmetrical. He is also the one who possesses the most acquired faculties. Among these faculties . . . the faculty of language holds pride of place. It is this that distinguishes us most clearly from the animals” (45) .
That Broca was right in supposing that directional asymmetry is the critical factor is now supported by studies of handedness in chimpanzees (46) and other primates (47) and studies of minicolumn separation in the planum temporale (48) . Directional asymmetries are absent in other primates, including the great apes, although there is some evidence that tool use in H. erectus was preferentially right-handed (49) .
Thus, to understand the human brain, it is necessary to understand the genetic basis of asymmetry (and its anatomical correlate—the cerebral “torque” from right frontal to left occipital). Two theories of handedness (50 , 51) postulate a single gene acting together with a random factor. Although both originally predicted an autosomal location, there is an association between handedness and sex within families (52) that is consistent with the presence of the gene on both X and Y chromosomes. Earlier, a gene in a region of X-Y homology was suggested on the basis of observations of sex chromosome aneuploidies (53) , and this is supported by intergenerational evidence from family studies (54 , 55) . The question remains: How could an apparently new feature—in this case cerebral asymmetry—arise from the existing genome complement? One hypothesis, championed, for example, by Goldschmidt (56) , an advocate of saltational change in evolution, is that chromosomal rearrangements play a role. Have there been chromosomal changes that might be relevant? One such change is the X to Y translocation followed by a paracentric inversion on the Y short arm that created a region of homology between Xq21.3 and two blocks on Yp after the separation of the chimpanzee from the hominid lineages (57 – 59) . These rearrangements ensured that gene sequences that are present on the X in all mammals are present in man on the Y chromosome also. The direction of the sequence on the Y was reversed in the original translocation (estimated to have taken place approximately 6 million years ago—that is, close to the time of, and maybe the cause of, separation of the chimpanzee and hominid lineages) and in the paracentric inversion was reversed again in the distal block in Yp ( Figure 3 ). Because the arrangement of the homologous regions on the Y is universal in modern human males, these changes must have been selected (i.e., the gene content of the block duplicated on the Y is relevant), and the chromosomal changes constitute speciation events in hominid evolution. The first rearrangement is a possible correlate of the transition from the chimpanzee-hominid precursor to Australopithecus , and the second (the paracentric inversion which cannot yet be dated) is a possible correlate of the transition to modern H. sapiens . Within this region of homology, a gene pair (protocadherin X/Y) coding for cell surface adhesion molecules has been identified (60) that has been subject to accelerated evolution (16 amino acid changes in the Y and five in the X sequence) in the hominid lineage by contrast with the relative stability of the sequence on the X in earlier primate evolution. This gene pair therefore is a candidate determinant of cerebral dominance for language.
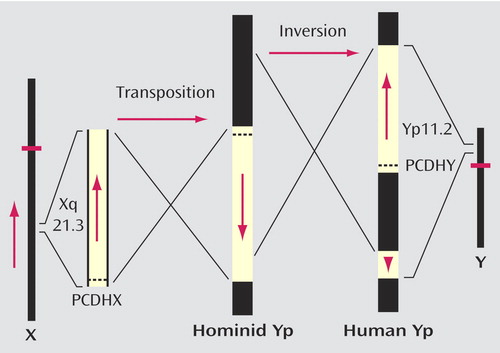
a The diagram shows a reduplicative transposition dated to approximately 6 million years ago and a subsequent (and at present undated) paracentric inversion that split the translocated block on the Y short arm of an intermediate hominid species. Adapted from Schwartz et al. (59; see also references 57 and 75) to show the location of the protocadherin X/Y gene pair (PCDHX and PCDHY). The original translocation is a possible speciation event in the transition from a great ape-hominid precursor to Australopithecus , and the paracentric inversion has a possible role in relation to the transition to H. sapiens . Note that the direction of the PCDHY sequence on Yp has possible relevance to the inactivation status of PCDHX in females (61).
Of particular interest is the status of protocadherin X/Y (and other genes within this region) with respect to X inactivation. Genes that are also present on the Y chromosome are protected from this epigenetic process, a protection that can be understood as aimed at achieving gene dosage equivalence in males and females. Therefore, whereas in all other mammals protocadherin X may be expected to be inactive on the inactive X chromosome, in females in H. sapiens a new situation pertains. The mechanism by which protection from X inactivation is brought about is obscure, but it must be supposed to depend on an interaction between X and Y chromosomes, and this can only take place in male meiosis (61) . If it depends on pairing of X-Y homologous sequences, then clearly the direction of the sequence on the Y and therefore the paracentric inversion will have significance. Whatever influence the presence of protocadherin Y may have had on the inactivation status of protocadherin X before the paracentric inversion, that influence will have been greater after this event. Thus, the orientation of the gene sequence on the Y must be supposed to have a key influence on the balance of gene expression in the two sexes in modern H. sapiens relative to earlier hominid species.
A Solution to the Problem of Psychosis?
Conventional wisdom is that the genes for psychosis are autosomal, but no such genes have been found. There are sex differences to be explained; the earliest onsets are predominantly in males and late onsets in females. There is also a same-sex concordance effect—a tendency for affected pairs of relatives (e.g., siblings) to be more often of the same sex than would be expected by chance (62 , 63) . This effect, noted by Penrose (64) and Rosenthal (65) , is predicted for a gene present on both X and Y chromosomes. Interestingly, the effect is at least as strong in affective as in schizophrenic psychoses (66) . There is evidence that the structural changes in schizophrenia interact with asymmetry and sex (67 – 69) . Thus it is plausible that the primary change lies in the variation associated with an X- and Y-linked determinant of the relative rates of development of the two hemispheres. (The X chromosome was omitted from Badner and Gershon’s meta-analysis of bipolar disorder [13] , Lewis et al.’s meta-analysis of schizophrenia [14] , and Segurado et al.’s meta-analysis of bipolar disorder [15] .) If, as argued here, the variation relating to psychosis is epigenetic, then this variation will be expected to arise in meiosis, the point at which a preexisting imprint is removed and a new one, depending on the interaction of the grandpaternal and grandmaternal Xs in female meiosis and the grandpaternal Y and the grandmaternal X in male meiosis, is imposed. In each case the interaction may be assumed to be a function of individual variations in DNA structure in adjacent genomic sequences. Because such sequences are shared by monozygotic twins more than by dizygotic twins and siblings, increased rates of concordance for psychosis will be expected in the former, as observed in the twin studies (9) notwithstanding that linkage is absent.
Given the generation of the Xq21.3/Yp regions of homology in the course of hominid evolution and the nature of the gene pair’s protein products as pathfinder molecules, protocadherins X and Y must be considered leading candidates. Sequence variation in this gene pair between individuals is low and has been shown to be unrelated to psychosis (70) . The epigenetic status of this pair and other genes within the transposed region thus becomes a focus of interest (71) . The fact that monozygotic twins often differ for handedness and its structural correlate, asymmetry of the planum temporale (72) , suggests that for the development of cerebral dominance for language, epigenetic factors are of primary significance in determining individual differences. The alternative to the search for the multiple genes of small effect that are so elusive to the linkage approach therefore is that the relevant variation is epigenetic and that it relates to the most characteristically human dimension—hemispheric dominance for language.
1. Sherrington R, Brynjolffson J, Petursson H, Potter M, Dudleston K, Barraclough B, Wasmuth J, Dobbs M, Gurling H: Localization of a susceptibility locus for schizophrenia on chromosome 5. Nature 1988; 336:164–167Google Scholar
2. Kennedy JL, Giuffra LA, Moises HW, Baillie D, Hubbard A, Wright A: Evidence against linkage of schizophrenia to markers on chromosome 5 in a northern Swedish pedigree. Nature 1988; 336:167–170Google Scholar
3. St Clair D, Blackwood D, Muir W, Baillie D, Hubbard A, Wright A, Evans HJ: No linkage of chromosome 5q11-q13 markers to schizophrenia in Scottish families. Nature 1989; 339:305–309Google Scholar
4. McGuffin P, Sargeant M, Hetti S, Tidmarsh S, Whatley S, Marchbanks RM: Exclusion of a schizophrenia susceptibility gene from the chromosome 5q11-q13 region: new data and a reanalysis of previous reports. Am J Hum Genet 1990; 47:524–535Google Scholar
5. Aschauer HN, Aschauer-Treiber G, Isenberg KE, Todd RD, Knesevich MA, Garver DL, Reich T, Cloninger CR: No evidence for linkage between chromosome 5 markers and schizophrenia. Hum Hered 1990; 40:109–115Google Scholar
6. Egeland JA, Gerhard DS, Pauls DL, Sussex JN, Kidd KK, Allen CR, Hostetter AM, Housman DE: Bipolar affective disorders linked to DNA markers on chromsome 11. Nature 1987; 325:783–787Google Scholar
7. Kelsoe JR, Ginns EI, Egeland JA, Gerhard DS, Goldstein AM, Bale SJ, Pauls DL, Long RT, Kidd KK, Conte G, Housman DE, Paul SM: Re-evaluation of the linkage relationship between chromosome 11p loci and the gene for bipolar affective disorder in the Old Order Amish. Nature 1989; 342:238–243Google Scholar
8. Baron M, Risch N, Hamburger R, Mandel B, Kushner S, Newman M, Drumer D, Belmaker RH: Genetic linkage between X chromosome markers and bipolar affective illness. Nature 1987; 326:289–292Google Scholar
9. Gottesman II: Schizophrenia Genesis: The Origins of Madness. New York, Freeman, 1991Google Scholar
10. Gershon ES: Genetics, in Manic-Depressive Illness. Edited by Goodwin FK, Jamison KR. New York, Oxford University Press, 1990, pp 373–401Google Scholar
11. Brzustowicz LH, Hodgkinson KA, Chow EWC, Honer WG, Bassett AS: Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science 2000; 288:678–682Google Scholar
12. Levinson DF, Holmans PA, Laurent C, Riley B, Pulver AE, Gejman PV, Schwab SG, Williams NM, Owen MJ, Wildenauer DB, Sanders AR, Nestadt G, Mowry BJ, Wormley B, Bauche S, Soubigou S, Ribble R, Nertney DA, Liang KY, Martinolich L, Maier W, Norton N, Williams H, Albus M, Carpenter EB, DeMarchi N, Ewen-White KR, Lerer B, O’Donovan MC, Dikeos D, Silverman JM, Kendler KS, Mallet J, Crowe RR, Walters M: No major schizophrenia locus detected on chromosome 1q in a larger multicenter sample. Science 2002; 296:739–741Google Scholar
13. Badner J, Gershon ES: Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry 2002; 7:405–411Google Scholar
14. Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LH, Kaufman CA, Garver DL: Genome scan meta-analysis of schizophrenia and bipolar disorder, II: schizophrenia. Am J Hum Genet 2003; 73:34–48Google Scholar
15. Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI Jr, et al: Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: bipolar disorder. Am J Hum Genet 2003; 73:49–62Google Scholar
16. Levinson DF, Levinson MD, Segurado R, Lewis CM: Genome scan meta-analysis of schizophrenia and bipolar disorder: methods and power analysis. Am J Hum Genet 2003; 73:17–33Google Scholar
17. DeLisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, Wellman N, Loftus J, Nanthakumar B, Razi K, Stewart J, Comazzi M, Vita A, Heffner T, Sherrington R: A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry 2002; 159:803–812Google Scholar
18. Stassen HH, Scharfetter C, Winokur G, Angst J: Familial syndrome patterns in schizophrenia, schizoaffective disorder, mania, and depression. Eur Arch Psychiatry Neurol Sci 1988; 237:115–123Google Scholar
19. Gershon ES, DeLisi LE, Hamovit J, Nurnberger JI, Maxwell ME, Schreiber J, Dauphinais D, Dingman CW, Guroff JJ: A controlled family study of chronic psychoses: schizophrenia and schizo-affective disorder. Arch Gen Psychiatry 1988; 45:328–336Google Scholar
20. Maier W, Lichtermann D, Minges J, Hallmayer J, Henn R, Benkert O, Levinson DF: Continuity and discontinuity of affective disorders and schizophrenia. Arch Gen Psychiatry 1993; 50:871–883Google Scholar
21. Kendler KS, McGuire M, Gruenberg AM, O’Hare A, Spellman M, Walsh D: The Roscommon Family Study, I: methods, diagnosis of probands, and risk of schizophrenia in relatives. Arch Gen Psychiatry 1993; 50:527–540Google Scholar
22. Williams NM, Norton N, Williams H, Ekholm B, Hamshere ML, Lindblom Y, Chowdari KV, Cardno AG, Zammit S, Jones LA, Murphy KC, Sanders RD, McCarthy G, Gray MY, Jones G, Holmans P, Nimgaonkar V, Osby U, Terenius L, Sedvall G, O’Donovan MC, Owen MJ: A systematic genomewide linkage study in 353 sib pairs with schizophrenia. Am J Hum Genet 2003; 73:1355–1367Google Scholar
23. Suarez BK, Duan J, Sanders AR, Hinrichs AL, Jin CH, Hou C, Buccola NG, Hale N, Weilbaecher AN, Nertney DA, Olincy A, Green S, Schaffer AW, Smith CJ, Hannah DE, Rice JP, Cox NJ, Martinez M, Mowry BJ, Amin F, Silverman JM, Black DW, Byerley WF, Crowe RR, Freedman R, Cloninger CR, Levinson DF, Gejman PV: Genomewide linkage scan of 409 European-ancestry and African American families with schizophrenia: suggestive evidence of linkage at 8p23.3-p21.2 and 11p13.1-q14.1 in the combined sample. Am J Hum Genet 2006; 78:315–333Google Scholar
24. Lambert D, Middle F, Hamshere ML, Segurado R, Raybould R, Corvin A, Green E, O’Mahony E, Nokolov I, Mulcahy T, Haque S, Bort S, Bennett P, Norton N, Owen MJ, Kirov G, Lendon C, Jones L, Jones I, Holmans P, Gill M, Craddock N: Stage 2 of the Wellcome Trust UK-Irish bipolar affective disorder sibling-pair genome screen: evidence for linkage on chromosomes 6q16-q21, 4q12-q21, 9p21, 10p14-p12, and 18q22. Mol Psychiatry 2005; 10:831–841Google Scholar
25. Harrison PJ, Owen MJ: Genes for schizophrenia? recent findings and their pathophysiological implications. Lancet 2003; 361:417–419Google Scholar
26. Duan J, Martinez M, Sanders AR, Hou C, Krasner AJ, Schwartz DB, Gejman PV: Neuregulin 1 (NRG1) and schizophrenia: analysis of a US family sample and the evidence in the balance. Psychol Med 2005; 35:1599–1610Google Scholar
27. DeLisi LE, Mesen A, Rodriguez C, Bertheau A, LaPrade M, Riondet S, Razi K, Relja M, Byerley W, Sherrington R: Genome-wide scan for linkage to schizophrenia in a Spanish-origin cohort from Costa Rica. Am J Med Genet 2002; 114:497–508Google Scholar
28. Torrey EF, Bowler AE, Taylor EH, Gottesman II: Schizophrenia and Manic-Depressive Disorder. New York, Basic Books, 1994Google Scholar
29. Risch N: Linkage strategies for genetically complex traits, 1: multilocus models. Am J Hum Genet 1990; 46:222–228Google Scholar
30. Crow TJ: A continuum of psychosis, one human gene, and not much else: the case for homogeneity. Schizophr Res 1995; 17:135–145Google Scholar
31. Johnstone EC, Crow TJ, Frith CD, Husband J, Kreel L: Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet 1976; 2:924–926Google Scholar
32. Vita A, Dieci M, Silenzi C, Tenconi F, Giobbio GM, Invernizzi G: Cerebral ventricular enlargement as a generalized feature of schizophrenia: a distribution analysis on 502 subjects. Schizophr Res 2000; 44:25–34Google Scholar
33. Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, Day R, Bertelsen A: Schizophrenia: manifestations, incidence, and course in different cultures: a World Health Organization ten-country study. Psychol Med Monogr Suppl 1992; 20:1–97Google Scholar
34. Weissman MM, Bland RC, Canino GJ, Faravelli C, Greenwald S, Hwu HG, Joyce PR, Karam EG, Lee CK, Lellouch J, Lepine JP, Newman SC, Rubio-Stipec M, Wells JE, Wickramaratne PJ, Wittchen H, Yeh EK: Cross-national epidemiology of major depression and bipolar disorder. JAMA 1996; 276:293–299Google Scholar
35. Esiri MM, Crow TJ: The neuropathology of psychiatric disorder, in Greenfield’s Neuropathology, 7th ed. Edited by Lantos PL, Graham DI. London, Arnold, 2002, pp 431–470Google Scholar
36. Pritchard DJ: Genetic analysis of schizophrenia as an example of a putative multifactorial trait. Ann Hum Genet 1996; 60:105–123Google Scholar
37. Karlsson JL: Inheritance of schizophrenia. Acta Psychiatr Scand Suppl 1974; 274:1–116Google Scholar
38. Crow TJ: Nuclear schizophrenic symptoms as a window on the relationship between thought and speech. Br J Psychiatry 1998; 173:303–309Google Scholar
39. Crow TJ: Auditory hallucinations as primary disorders of syntax: an evolutionary theory of the origins of language. Cognit Neuropsychiatry 2004; 9:125–145Google Scholar
40. Stringer C, McKie R: African Exodus: The Origins of Modern Humanity. London, Cape, 1996Google Scholar
41. Noble W, Davidson I: Human Evolution, Language, and Mind. Cambridge, UK, Cambridge University Press, 1996Google Scholar
42. Mellars P: Archaeology and the origins of modern humans: European and African perspectives, in The Speciation of Modern Homo Sapiens. Edited by Crow TJ. Oxford, UK, Oxford University Press, 2002, pp 31–48Google Scholar
43. Broca P: Remarques sur le siège de la faculté du langage articulé, suivies d’une observation d’aphémie (perte de la parole). Bull Soc Anat Paris 1861; 6:330–357Google Scholar
44. Dax M: Lésions de la moitié gauche de l’éncephale coincident avec l’oubli des signes de la pensée. Gaz Hebdom Méd Chirurg 1865; 11:259–260Google Scholar
45. Broca P: Rapport sur un mémoire de M. Armand de Fleury intitulé: De l’inégalité dynamique des deux hémisphères cérébraux. Bull Acad Médicine 1877; 6:508–539Google Scholar
46. Marchant LF, McGrew WC: Laterality of limb function in wild chimpanzees of Gombe National Park: comprehensive study of spontaneous activities. J Hum Evol 1996; 30:427–443Google Scholar
47. Holder MK: Influences and constraints on manual asymmetry in wild African primates: reassessing implications for the evolution of human handedness and brain lateralization. Dissertation Abstracts International 1999; 60(A):0470Google Scholar
48. Buxhoeveden D, Switala AE, Litaker M, Roy E, Casanova MF: Lateralization of minicolumns in human planum temporale is absent in nonhuman primate cortex. Brain Behav Evol 2001; 57:349–358Google Scholar
49. Steele J: When did directional asymmetry enter the record? in The Speciation of Modern Homo Sapiens. Edited by Crow TJ. Oxford, UK, Oxford University Press, 2002, pp 153–168Google Scholar
50. Annett M: Left, Right, Hand, and Brain: The Right Shift Theory. London, Erlbaum, 1985Google Scholar
51. McManus IC: Handedness, language dominance, and aphasia: a genetic model. Psychol Med Monogr Suppl 1985; 8:1–40Google Scholar
52. Corballis MC, Lee K, McManus IC, Crow TJ: Location of the handedness gene on the X and Y chromosomes. Am J Med Genet 1996; 67:50–52Google Scholar
53. Crow TJ: Sexual selection, Machiavellian intelligence, and the origins of psychosis. Lancet 1993; 342:594–598Google Scholar
54. Jones GV, Martin M: A note on Corballis (1997) and the genetics and evolution of handedness: developing a unified distribution model from the sex-chromosomes gene hypothesis. Psychol Rev 2000; 107:213–218Google Scholar
55. Jones GV, Martin M: Confirming the X-linked handedness gene as recessive, not additive: reply to Corballis (2001). Psychol Rev 2001; 108:811–813Google Scholar
56. Goldschmidt R: The Material Basis of Evolution. New Haven, Conn, Yale University Press, 1940Google Scholar
57. Sargent CA, Briggs H, Chalmers IJ, Lambson B, Walker E, Affara NA: The sequence organization of Yp/proximal Xq homologous regions of the human sex chromosomes is highly conserved. Genomics 1996; 32:200–209Google Scholar
58. Mumm S, Molini B, Terrell J, Srivastava A, Schlessinger D: Evolutionary features of the 4Mb Xq21.3 XY homology region revealed by a map at 60-kb resolution. Genome Res 1997; 7:307–314Google Scholar
59. Schwartz A, Chan DC, Brown LG, Alagappan R, Pettay D, Disteche C, McGillivray B, de la Chapelle A, Page DC: Reconstructing hominid Y evolution: X-homologous block, created by X-Y transposition, was disrupted by Yp inversion through LINE-LINE recombination. Hum Mol Genet 1998; 7:1–11Google Scholar
60. Williams NA, Close J, Giouzeli M, Crow TJ: Accelerated evolution of Protocadherin11xy: a candidate gene-pair for cerebral asymmetry and language. Am J Med Genetic B Neuropsychiatr Genet 2006; 141:623–633Google Scholar
61. Crow TJ: Protection from X inactivation. Nature 1991; 353:710Google Scholar
62. Crow TJ, DeLisi LE, Johnstone EC: Concordance by sex in sibling pairs with schizophrenia is paternally inherited: evidence for a pseudoautosomal locus. Br J Psychiatry 1989; 155:92–97Google Scholar
63. Crow TJ, DeLisi LE, Johnstone EC: In reply: a locus closer to the telomere? Br J Psychiatry 1990; 156:416–420Google Scholar
64. Penrose LS: Auxiliary genes for determining sex as contributory causes of mental illness. J Ment Sci 1942; 88:308–316Google Scholar
65. Rosenthal D: Familial concordance by sex with respect to schizophrenia. Psychol Bull 1962; 59:401–421Google Scholar
66. Crow TJ: Con: The demise of the Kraepelinian binary system as a prelude to genetic advance, in Genetic Approaches to Mental Disorders. Edited by Gershon ES, Cloninger CR. Washington, DC, American Psychiatric Press, 1994, pp 163–192Google Scholar
67. Crow TJ: Temporal lobe asymmetries as the key to the etiology of schizophrenia. Schizophr Bull 1990; 16:433–443Google Scholar
68. Highley JR, McDonald B, Walker MA, Esiri MM, Crow TJ: Schizophrenia and temporal lobe asymmetry: a post mortem stereological study of tissue volume. Br J Psychiatry 1999; 175:127–134Google Scholar
69. McDonald B, Highley JR, Walker MA, Herron BM, Cooper SJ, Esiri MM, Crow TJ: Anomalous asymmetry of fusiform and parahippocampal gyrus gray matter in schizophrenia: a postmortem study. Am J Psychiatry 2000; 157:40–47Google Scholar
70. Giouzeli M, Williams NA, Lonie L, DeLisi LE, Crow TJ: Protocadherin X/Y, a candidate gene-pair for schizophrenia and schizoaffective disorder: a DHPLC investigation of genomic sequence. Am J Med Genet B Neuropsychiatr Genet 2004; 129:1–9Google Scholar
71. Crow TJ: Handedness, language lateralisation, and anatomical asymmetry: relevance of protocadherin XY to hominid speciation and the aetiology of psychosis. Br J Psychiatry 2002; 181:295–297Google Scholar
72. Steinmetz H, Herzog A, Schlaug G, Huang Y, Jancke L: Brain (a)symmetry in monozygotic twins. Cereb Cortex 1995; 5:296–300Google Scholar
73. Bickerton D: Language and Human Behavior. Seattle, University of Washington, 1995Google Scholar
74. Collard M: Grades and transitions in human evolution, in The Speciation of Modern Homo Sapiens. Edited by Crow TJ. Oxford, UK, Oxford University Press, 2002, pp 61–102Google Scholar
75. Sargent CA, Blanco P, Affara NA: Do the hominid specific regions of X-Y homology contain candidate genes potentially involved in a critical event linked to speciation? in The Speciation of Modern Homo Sapiens. Edited by Crow TJ. Oxford, UK, Oxford University Press, 2002, pp 231–250Google Scholar

