
Revisiting Schizophrenia Linkage Data in the NIMH Repository: Reanalysis of Regularized Data Across Multiple Studies
Abstract
Objective
The Combined Analysis of Psychiatric Studies (CAPS) project conducted extensive review and regularization across studies of all schizophrenia linkage data available as of 2011 from the National Institute of Mental Health-funded Center for Collaborative Genomic Studies on Mental Disorders, also known as the Human Genetics Initiative (HGI). The authors reanalyzed the data using statistical methods tailored to accumulation of evidence across multiple, potentially highly heterogeneous, sets of data.
Method
Data were subdivided based on contributing study, major population group, and presence or absence within families of schizophrenia with a substantial affective component. The posterior probability of linkage (PPL) statistical framework was used to sequentially update linkage evidence across these data subsets (omnibus results).
Results
While some loci previously implicated using the HGI data were also identified in the present omnibus analysis (2q36.1, 15q23), others were not. Several loci were found that had not previously been reported in the HGI samples but are supported by independent linkage or association studies (3q28, 12q23.1, 11p11.2, Xq26.1). Not surprisingly, differences were seen across population groups. Of particular interest are signals on 11p15.3, 11p11.2, and Xq26.1, for which data from families with a substantial affective component support linkage while data from the remaining families provide evidence against linkage. All three of these loci overlap with loci reported in independent studies of bipolar disorder or mixed bipolar-schizophrenia samples.
Conclusions
Public data repositories provide the opportunity to leverage large multisite data sets for studying complex disorders. Analysis with a statistical method specifically designed for such data enables us to extract new information from an existing data resource.
For the past two decades, investigators funded by the National Institute of Mental Health (NIMH) conducting genetic research have been strongly encouraged to contribute biospecimens, along with whatever corresponding genotypic and phenotypic information they have assembled, to a centralized repository housed at the Center for Collaborative Genomic Studies on Mental Disorders. The repository grows immortalized cell lines, supplies DNA to researchers, and provides downloadable copies of clinical and genotypic data files through the Human Genetics Initiative (HGI) (see www.nimhgenetics.org). We report the first wave of results from the HGI’s Combined Analysis of Psychiatric Studies (CAPS) project, which is undertaking extensive review and analysis of data in the repository. Our focus here is the reanalysis of the seven separate schizophrenia family studies that had deposited genotypic and phenotypic data with the repository as of April 2011.
The CAPS project is specifically funded to reanalyze HGI data using the posterior probability of linkage (PPL) framework as implemented in the software package KELVIN (1). There are three principal advantages to using the PPL for linkage analysis of the HGI data (see the Method section for details). First, it is essentially model free, while retaining the advantages of likelihood-based analyses, making full use of all available data, including from unaffected individuals. Second, it is specifically tailored to handle multiple, potentially highly heterogeneous data sets or subsets, using Bayesian sequential updating to accumulate linkage evidence across subsets while allowing explicitly for genetic differences between subsets. And third, in stark contrast to p values or maximum LOD scores, based on either “mega-analysis” or traditional meta-analysis, the PPL can accumulate evidence both for and against linkage at each genomic position. This increases resolution of a genome scan by excluding stretches of the genome in an essentially model-free manner, while also distinguishing common as opposed to distinct genetic features across different populations or clinical subgroups.
The PPL is thus uniquely well suited to the analysis of multisite, potentially highly heterogeneous, genetic data. Here we consider omnibus linkage results, based on all of the HGI schizophrenia family data in aggregate. We also consider population-specific findings and the possibility of specific loci underlying a clinical schizophrenia subtype involving affective components.
Method
Data Classification and Subdivision
Data for all multiplex pedigrees with available genome-wide genotyping available as of April 2011 were downloaded from the HGI. Families in the download came from seven different studies (2–9), referred to here as studies 1–7. An extensive data regularization protocol was applied to both genotypic and phenotypic information as downloaded from the HGI. This included applying strict criteria to the DSM diagnostic codes provided to the HGI by the individual studies to define a narrow schizophrenia phenotype and a schizophrenia/affective phenotype, which included schizoaffective disorder or any schizophrenia disorder and a significant affective disorder. (See online data supplements SA1 and SA2 for details of genotype and phenotype processing, respectively.)
Families were included in these analyses if they contained at least one case of schizophrenia, at least one additional case of either schizophrenia or schizophrenia/affective diagnoses, and at least two affected genotyped individuals. This left 970 multiplex families for analysis, containing 4,023 phenotyped individuals (average, 4.1 per pedigree) and 4,208 genotyped individuals (average, 4.3 per pedigree). (See online data supplement ST1 for an annotated list of families included and excluded from these analyses.) We elected to use stringent diagnostic and inclusion criteria in order to minimize clinical heterogeneity within data subsets, and especially in order to minimize the effects of clinical differences across studies. Such differences may tend to be more pronounced for broad-spectrum conditions than for the core diagnoses.
We grouped families into subsets by study and population (African American, European American, Han Chinese, and Hispanic). Each of the original studies either contained a single group or, in the cases of study 1 (2, 3) and study 2 (4), divided their own analyses into European American and African American families in published analyses. Thus, these subgroupings match previous treatments of the data sets.
Additionally, because schizophrenia with a significant affective component is now considered to be a potentially genetically distinct form of schizophrenia, we further subdivided the data based on the presence or absence of any individuals with schizophrenia/affective diagnoses within the pedigree. (When referring to families, “schizophrenia” refers to families whose affected members are all classified as having schizophrenia; and “schizophrenia/affective” refers to families in which at least one affected individual is classified as having a schizophrenia/affective diagnosis.) While it is impossible to distinguish a family that would never have produced a case meeting schizophrenia/affective criteria from a family that, by chance, failed to manifest this affective phenotype among the small number of affected individuals present, even an imperfect classifier can be helpful in detecting linkage in one but not the other group (10).
Several of the studies had multiple data collection sites, and “site” itself might demarcate more homogeneous subsets of the data. However, further subdivision by site results in sample sizes that are too small for meaningful analysis (<10 small families/data set), so we did not subdivide by site. (See online data supplement ST2 for sample sizes by subset.)
Statistical Analysis
All analyses were conducted using the software package KELVIN (1), which implements the PPL class of models for measuring the strength of genetic evidence. The specific statistic employed here was the PPL itself (posterior probability of linkage). The PPL is essentially a straightforward application of Bayes’s theorem. Letting L represent “linkage” to a given genomic position and D be the data, the PPL is calculated as The two likelihoods appearing in this equation, P(D|L) and P(D|no L), are the numerator and denominator, respectively, of the exponentiated ordinary LOD score. They are functions of the parameters of the unknown trait model, and what distinguishes the PPL as computed by KELVIN is the way in which these parameters are handled within and across data subsets, as described below.
The two likelihoods appearing in this equation, P(D|L) and P(D|no L), are the numerator and denominator, respectively, of the exponentiated ordinary LOD score. They are functions of the parameters of the unknown trait model, and what distinguishes the PPL as computed by KELVIN is the way in which these parameters are handled within and across data subsets, as described below.
The PPL as applied here represents the probability of a schizophrenia gene at each location, given the available data. For the microsatellite data, four-point (three-marker) multipoint calculations were run; for the single-nucleotide polymorphism data, full multipoint analysis using all markers was used. (Technical details and all primary references to the supporting theoretical literature may be found in reference 1; examples of previous applications may be found in references 11–13.)
The PPL is based on an approximating single-locus likelihood allowing for within-sample heterogeneity using Smith’s admixture parameter α (14). All parameters of the trait model, including α, are integrated out of the PPL using (independent) uniform priors on each parameter (see reference 1 for details), implicitly allowing for dominant, recessive, or intermediate models. This provides a robust approximation for mapping complex traits in terms of the marginal model at each locus, and because the parameters are integrated out, no specific assumptions regarding their values are required.
The PPL framework accumulates evidence across data subsets by integrating the trait parameters out of the likelihood separately for each subset (allowing their values to differ across subsets), then using Bayesian sequential updating to carry the posterior linkage evidence from previously analyzed data forward as prior evidence as new data subsets are analyzed at each genomic position in turn. This allows for differences in allele frequencies, penetrances (due to background genetic or environmental differences across subsets), and chance fluctuations in the proportion of “linked” pedigrees at a given locus. The order in which data sets are analyzed does not affect the final result. In the presence of appreciable heterogeneity, sequential updating is far more robust in retaining true signals originating from individual subsamples than analyses that simply combine subsets for a single analysis (10, 15, 16). It is also substantially more sensitive in detecting true genetic effects across heterogeneous disorders than standard meta-analyses (17, 18). Moreover, the PPL accumulates evidence against linkage as well as in favor of linkage. Thus, inspection of subset-specific contributions to the omnibus signal can distinguish among subsets that are (perhaps only weakly) supporting linkage and subsets that are actually contributing evidence against linkage, as we illustrate below. Here we sequentially updated across data subsets defined by study (studies 1–7), major population group (African American, European American, Han Chinese, Hispanic), and clinical type (presence or absence of any individual meeting schizophrenia/affective criteria within the family).
The PPL is on the probability scale, and its interpretation is therefore straightforward: e.g., a PPL of 40% means that there is an estimated 40% probability of a trait gene at the given location based on these data. Based on Elston and Lange’s analysis (19), the prior probability at each location is set to 2%, so that PPLs >2% indicate (some degree of) evidence in favor of a trait gene at the locus, while PPLs <2% represent evidence against the location. This prior probability is based on empirical data (19), and because it is calculated as the probability of linkage between a random marker location and a single disease gene, it is conservative for multilocus disorders. The PPL is a measure of statistical evidence, not a decision-making procedure; therefore, there are no “significance levels” associated with it, and it is not interpreted in terms of associated error probabilities (20, 21). By the same token, no multiple testing corrections are applied to the PPL, just as one would not “correct” a measure of the temperature made in one location for readings taken at different locations (22). Nevertheless, it may assist readers to have some sense of scale relative to more familiar frequentist test statistics. In simulations of 10,000 replicates of sets of 1,000 affected sib pairs (the predominant data structure in these analyses) under the null hypothesis (no linkage), “significance” cutoffs of PPLs of 5%, 10%, 15%, and 25% were associated with type I error probabilities (the rate at which the PPL crossed the given threshold under the null) of 0.0070, 0.0011, 0.0004, and 0.0001, respectively. In the Results section, we highlight loci with PPL ≥25%; complete results are presented in online data supplement ST3.
Results
We first present omnibus linkage results, considering all of the data in aggregate. We then consider common and distinct loci, by major population group and by clinical subgroup (families with narrow schizophrenia only versus families containing at least one case of a schizophrenia/affective diagnosis). We then compare our results with results from the original publications for studies 1–7, as well as with results based on independent sets of data and meta-analyses.
Omnibus Results
Figure 1 presents omnibus linkage results, sequentially updated across all data subsets. Overall, 76% of the genome showed evidence against linkage (PPL<0.02), while only 10% showed PPL>0.05 and 4% showed PPL>0.1. There were twenty distinct loci with PPL>0.1, 13 with PPL>0.15, and four with PPL>0.25. These last occurred at 2q36.1 (PPL=0.41), 3q28 (PPL=0.27), 12q23.1 (PPL=0.26), and 15q23 (PPL=0.27) (Table 1).
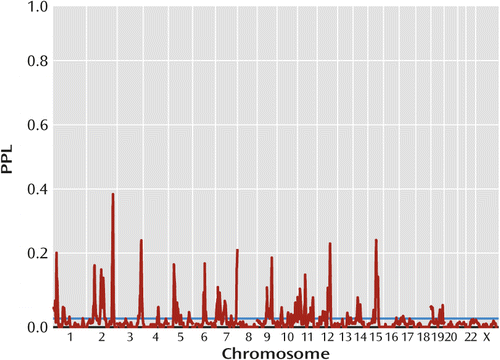
a Data from the Human Genetics Initiative. The posterior probability of linkage (PPL) is on the probability scale (0 to 1.0); values <2% (horizontal line) indicate evidence against linkage, while values >2% indicate (some degree of) evidence in favor of linkage.
| Locus | Genetic Position (cM)b | Omnibus Results | African American | European American | Han Chinese | Hispanic | Schizophrenia/Affective Families | Schizophrenia Families |
|---|---|---|---|---|---|---|---|---|
| 1p32.3 | 80 | 0.06 | 0.015 | 0.27 | 0.010 | 0.014 | 0.02 | 0.06 |
| 2q36.1 | 230 | 0.41 | 0.05 | 0.02 | 0.02 | 0.18 | 0.16 | 0.07 |
| 3q28 | 210 | 0.27 | 0.03 | 0.15 | 0.02 | 0.03 | 0.18 | 0.03 |
| 5p14.1 | 48 | 0.20 | 0.04 | 0.019 | 0.010 | 0.26 | 0.09 | 0.04 |
| 6q14.1–q15 | 94 | 0.10 | 0.017 | 0.11c | 0.018 | 0.03 | 0.000 | 0.46 |
| 10q22.3 | 98 | 0.04 | 0.015 | 0.009 | 0.36 | 0.009 | 0.009 | 0.11 |
| 10q26.12 | 144 | 0.04 | 0.010 | 0.008 | 0.34 | 0.015 | 0.010 | 0.09 |
| 11p15.3 | 22 | 0.12 | 0.10 | 0.02 | 0.015 | 0.04 | 0.30 | 0.009 |
| 11p11.2 | 68 | 0.14 | 0.06 | 0.09 | 0.014 | 0.016 | 0.36 | 0.008 |
| 12q23.1–q23.2 | 112 | 0.26 | 0.08c | 0.03 | 0.02 | 0.04 | 0.017 | 0.30c |
| 15q23 | 72 | 0.27 | 0.02 | 0.03 | 0.02 | 0.18 | 0.10 | 0.06 |
| Xq26.1 | 146 | 0.011 | 0.04 | 0.012 | 0.010 | 0.02 | 0.27 | 0.000 |
Population-Specific Results
Figure 2 presents population-specific results. There are four population-specific loci with PPL>0.25 (Table 1). Two of these come from Han Chinese (10q22.3, PPL=0.36; 10q26.12, PPL=0.34), and at both these loci all three of the other population groups show evidence against linkage. Similarly, European American data support 1p32.3 (PPL=0.27), with data from all other population groups providing evidence against linkage at this locus. By contrast, the Hispanic-specific peak at 5p14.1 (PPL=0.26) receives modest support from African American data (PPL=0.04) but evidence against linkage in the other two sets. The four omnibus peaks (see above) are each supported by at least three population groups, in each case with the remaining group being neutral, that is, showing evidence neither for nor against linkage (Table 1).
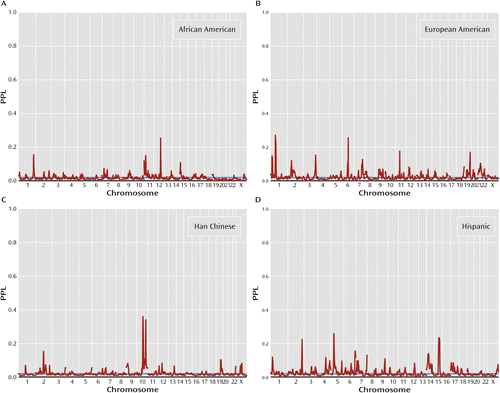
a Data from the Human Genetics Initiative. PPL=posterior probability of linkage.
Clinical Subset-Specific Results
Figure 3 presents results by clinical subgroup. There are five loci with PPL>0.25 in either of the two clinical subgroups (Table 1), and in every case the other subgroup provides evidence against linkage (in each case, PPL<0.01). Notably, three of these peaks (11p15.3, 11p11.2, 23q26.1) are supported by the far smaller schizophrenia/affective subset, suggesting that this really does constitute a more homogeneous classification of the families. However, there is possible confounding by population group here, since each clinical subset includes all four population groups, but in differing proportions (see the Discussion section).

a Data from the Human Genetics Initiative. PPL=posterior probability of linkage.
Comparison With Original Study Results
Figure 4 illustrates salient differences between the omnibus results and results extracted from the original published linkage analyses of the individual studies. The 10% PPL threshold was chosen based on qualitative visual separation of salient signals from background noise in Figure 1 and is intended to mimic the criterion of “suggestive” evidence as shown in the figure based on the original publications. There is no rigorous way to precisely compare the magnitude of results across data-analytic methods or to derive and apply exactly comparable interval estimates. Thus, the figure is intended to convey in broad strokes differences in the overall genomic landscape between the individual original reports and our omnibus analyses rather than to present precise quantitative differences.
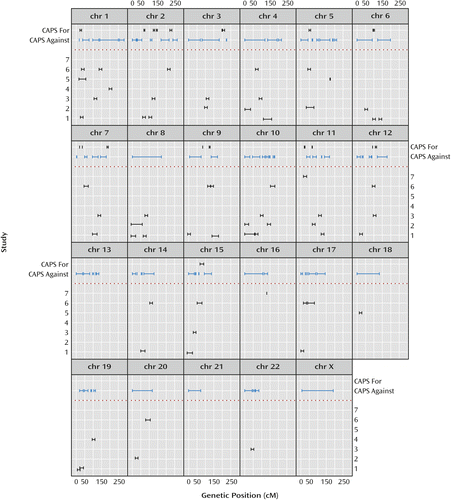
a Thresholds and intervals for inclusion of original signals were taken directly from published reports and generally correspond to “suggestive” linkage. Where intervals were not reported, the average reported interval of 16 cM was imposed. Omnibus posterior probability of linkage (PPL) results are shown above the dotted line. “CAPS For” includes all regions with PPL>10%. This threshold was selected to similarly reflect what might correspond to a “suggestive” level of support. “CAPS Against” includes all regions with PPL<2% (evidence against linkage). Note also that because of the low resolution of genetic scale in this figure, no attempt was made to harmonize the genetic maps across the original reports; different studies reported their results on Rutgers, Marshfield, Genethon, and genetic location database maps.
Of the 20 intervals with PPL>0.10, 12 line up in or near an originally reported interval. This could be coincidental, however, since all but three chromosomes are implicated (generally at the level of “suggestive” linkage) by at least one of the original publications, with little overlap in reported loci across the original studies. Figure 4 also shows the large portions of the genome showing evidence against linkage using the PPL. The PPL’s ability to accumulate evidence against linkage has no correlate in either the original nonparametric linkage or meta-analyses.
Comparison With Results From Independent Studies
Of the four omnibus loci with PPL over 25%, all are in regions of previous schizophrenia linkage reports. Chromosome 2q36.1 is in a region that has been implicated in several previous linkage reports, including a significant finding in a Finnish sample (23), and has received suggestive support from other reports (5, 24, 25) that include families found in study 2 and study 3. SCG2, the gene encoding secretogranin II, is located under the center of this peak and has been reported to have increased expression in the dorsolateral prefrontal cortex in individuals with schizophrenia (26). Chromosome 3q28 is supported by suggestive linkage results in studies in Finnish (23), Palauan (27), and Dagestani (28) samples that are not part of the NIMH-HGI collection. Chromosome 12q23.1 has been implicated by a suggestive linkage report with a negative symptom factor score (29). While the 12q candidate gene DAO is located beyond the boundaries of this peak, in an area where the omnibus analysis produced evidence against linkage, the gene PAH is located near the edge of the omnibus peak, in a region with PPL scores slightly above 2%. PAH, the gene for phenylalanine hydroxylase, while traditionally associated with phenylketonuria, has been suggested as a schizophrenia candidate gene in multiple studies (29–33). Of further interest is the 12q linkage peak observed in the African American subset; this peak has a maximum PPL score of 25.4% located 4 centimorgans (cM) telomeric to the omnibus peak, very near PAH. The omnibus peak on chromosome 15q23 overlays a suggestive linkage peak that was originally reported in the Hispanic samples included in this analysis (8).
Four additional peaks over 25% were observed in specific population groups. The 10q22.3 and 10q26.12 peaks in the Han Chinese sample overlap the linked regions from the original report on these families (5). Similarly, the Hispanic-specific peak at 5p14.1 overlaps a suggestive linkage peak that was previously reported in these families (8). By contrast, the 1p32.3 locus seen in the European American subset was not suggested by the individual studies contributing to this subset (4, 34, 35). Having data from one population group provide evidence for linkage and data from the other groups provide combined evidence against is consistent with either separate sets of genes operating in the different population groups or, more likely, dependence of the salience of genetic effects on the background (ancestral) genome or environment. It is also consistent, however, with the subset-specific PPLs being overestimates of the actual probability of linkage, which is given by the omnibus PPL. Differences in sample sizes across groups (139 African American, 187 European American, 483 Han Chinese, and 161 Hispanic) also complicate interpretation.
Dividing the data by clinical subgroup (families containing individuals with schizophrenia/affective diagnoses versus families with only narrow schizophrenia) yielded four additional loci with PPL>25%, and in every case the locus was supported by data from one group while data from the other group provided evidence against linkage. This strongly suggests that the genetics of schizophrenia in families characterized by the presence of schizoaffective disorder or schizophrenia with a comorbid affective disorder may be distinct from the genetics of schizophrenia in individuals without a significant mood disorder. On the other hand, there is considerable confounding by population, since the preponderance of families with individuals with a schizophrenia/affective diagnosis varies by group as well. The proportions of groups (African American, European American, Han Chinese, Hispanic) within schizophrenia-only families and families that include schizophrenia/affective diagnoses are 0.11, 0.15, 0.59, 0.15 and 0.28, 0.41, 0.04, 0.26, respectively.
Interestingly, however, all three locations identified in the families with schizophrenia/affective diagnoses (11p15.3, 11p11.2, and Xq26.1) have been previously implicated in linkage or association studies using samples with bipolar disorder or mixed samples with schizophrenia and bipolar disorder (36–40). The 11p15.3 region was also reported as a main finding in the original publication for study 7 (9), and indeed, the African American-specific PPL reaches 15% at 28 cM. Of particular note is that study 7 reported (9) a marked strengthening of the linkage signal at 11p15.3 when using a phenotype that included schizophrenia and schizoaffective disorder rather than schizophrenia only. Finally, the schizophrenia subset produced a peak of 46% at 6q14.1 that overlaps a peak of 26% in the European American subgroup but was not reported by any of the original European American studies. In aggregate, these results have significant implications for the inclusion of individuals with schizoaffective disorder in linkage or association studies of schizophrenia. (See also online data supplement SA3 for a comparison with results in the National Human Genome Research Institute genome-wide association study database.)
Insofar as population is a major source of heterogeneity in these analyses, it is noteworthy that, with the exception of the Han Chinese sample, the remaining sample sizes for the different groups remain quite small. It is not surprising, therefore, that even the largest PPLs remain moderate in size. Similarly, dividing the data based on the presence of affective illness results in a far smaller subset for the group containing schizophrenia/affective diagnoses. Strikingly, however, this group also produces some of the highest PPLs. Our results suggest that both population and clinical subgroup are key classifiers for the purposes of gene mapping. Ideally, we would like to use subsets simultaneously on both criteria. However, the extremely small subsets that would result preclude this approach.
It is also of interest to compare our results with the meta-analyses of Nato et al. (41) and Ng et al. (42), both of which included studies from the HGI collection as well as other studies. Nato et al. identified 22 regions of interest, while Ng et al. reported eight regions with aggregate genome-wide evidence for linkage, all of which overlap with regions identified by Nato et al. While the majority of the Nato et al. (13 of 22) and Ng et al. (five of eight) regions produced some level of evidence in favor of linkage under the omnibus analysis, only two of our four omnibus peaks >25% (2q36.1 and 3q28) overlapped a region of interest from either study, with our largest peak of 41% overlapping regions of interest from both studies. Of our eight peaks >25% that were specific to an ethnic or clinical subset, only two (5p14.1 and 10q26.12) overlapped a region of interest from either study, again with the larger peak of 34% overlapping regions of interest from both studies. (See online data supplement ST4 for detailed comparisons.)
Discussion
Our reanalyses of HGI data present a picture of the schizophrenia genome with some overlap with previous reports but also some novel loci, with differences both in terms of the specific loci implicated and in the rank ordering of these loci by relative strength of evidence. While a number of loci previously implicated using the samples from the HGI were also identified in the present analyses, such as 2q36.1 and 15q23, others were not. At the same time, several loci not previously reported in the HGI samples but supported by other reports in the literature have now been identified in the HGI collection, such as 3q28 and 12q23.1. Loci without any previous linkage reports in schizophrenia but with positional support from association studies were also identified, such as 11p11.2 and Xq26.1. Differences in the genetics of schizophrenia across population groups and in families with differing amounts of affective illness were also highlighted, in particular with 11p15.3, 11p11.2, and Xq26.1, which are prominent in the schizophrenia/affective group, overlapping with loci reported from independent studies of bipolar disorder or mixed bipolar-schizophrenia samples.
One caveat is that we employed a highly conservative approach both in deciding whom to categorize as “affected” (e.g., narrow schizophrenia with multiple exclusion criteria) and in our inclusion criteria for families (at least one case of schizophrenia plus one additional schizophrenia or schizophrenia/affective diagnosis), which led us to drop a large number of families from these analyses. While we believe that this approach is justified, particularly when attempting to make “apples to apples” comparisons across multiple different studies, we are aware that other investigators might make quite different judgments regarding these decisions. All data on the full set of families, including those we did not analyze here, are available on the HGI web site (www.nimhgenetics.org), as are all protocols used in preparing the data files (www.nimhgenetics.org/projects/CAPS). We hope that other investigators will find this a useful resource for carrying out their own analyses.
A second potential caveat is that we relied on linkage analysis. Given the relative paucity of validated linkage findings in psychiatric genetics, it is not unreasonable to be skeptical regarding this approach. There is a certain irony here, because past failures may be attributable in part to the fact that psychiatry was an early and enthusiastic endorser of the technique. Some of the schizophrenia data considered here date back to the 1990s, and the original studies taken individually were almost certainly underpowered, for a variety of reasons. By revisiting the HGI data and applying modern data processing and newer data-analytic methods to the existing multisite data as a whole, we believe that we have succeeded in extracting new information from older data sets of families. Of course, we have also lost something in working with the repository data, insofar as the original investigators had access to additional clinical information.
Historically there have been few gene discoveries under psychiatric linkage peaks. In this regard, too, having been a leader in methodological development put the field of psychiatric genetics ahead of its time: earlier molecular techniques were cumbersome and expensive, and combined with the width of linkage peaks in the older studies, this made gene identification under the peaks impractical. But the increasing practicality of high-throughput DNA sequencing should support gene discovery at linked loci in a manner unavailable until very recently. Indeed, human genetics is returning to a focus on “co-segregation” (i.e., linkage analysis) as a critical adjuvant to whole genome sequencing, because narrowing the search down to only those portions of the genome showing evidence of linkage dramatically reduces the amount of sequence needing to be interrogated. One could even argue that linkage analysis as a technique for gene discovery has yet to be properly applied in much of psychiatric genetics.
Even so, it might be argued that genome-wide association studies and case-control-based whole genome sequencing are a better investment than multiplex family studies. But linkage analysis and association analysis (whether to common single-nucleotide polymorphisms or rare variants) have the power to detect very different types of genetic effects: genome-wide association studies are well adapted to finding common variants conferring low relative risks in a manner that is consistent across populations (43) (requiring independent replication ensures that effects specific to one data set are discounted [44]). While linkage analysis is adapted to finding loci containing genes of large effect (in general, with rarer allele frequencies and much higher genotypic relative risks), and particularly when conducted using statistical methods specifically tuned for such conditions, it can find such genes even when they are causally relevant only in a subset of families (10). One reason to continue to search for such genes is precisely because a gene discovered by linkage analysis almost certainly is a gene of large effect, albeit perhaps only in some families. Such genes may yield different insights into gene pathways and networks than genes discovered through alternative study designs. Linkage analysis is also robust to allelic heterogeneity, which can eliminate allelic associations altogether (45).
The genetic architecture for complex disorders likely involves many different types of effects simultaneously: major gene effects in subsets of families; common background genes conferring small risks; copy number variants, perhaps particularly important in causing sporadic forms of disease; extensive locus heterogeneity, which might or might not be alleviated by refined clinical subtyping; and more complex features, including gene-by-gene and gene-by-environment interactions, epigenetics, and probably other things we have not even thought of yet. Under such circumstances, no one study design can solve all problems.
The psychiatric genetics community has an enormous resource on hand: extensive collections of multiplex families with considerable clinical information. These collections were labor intensive and expensive to amass; by contrast, the cost of genotyping or even sequencing is becoming relatively small. Returning to these collections with the benefits of hindsight seems a promising and cost-effective way to contribute to the growing understanding of genetic architecture emerging from a multitude of studies and study designs all being considered simultaneously. We have tried here to illustrate the potential of such use of retrospective data to alter and augment our understanding of the genetic underpinnings of psychiatric disorders. Of course, interpretation of the results is still hampered by the limited information content afforded by the available genotype data.
Finally, we note an important methodological issue highlighted by these results, namely, that conventional interpretations of statistical significance in terms of independent replication can lead us to overlook important loci that may have salient effects only in subsets of the data. As is well known, failure to replicate true loci is to be expected for even moderately complex disorders (46). What is perhaps less widely appreciated is that traditional meta-analysis will tend to fail in precisely those same circumstances where independent replication cannot be relied upon for confirmation of results (17, 18). At the same time, we are appropriately skeptical of weak findings that fail to replicate. Indeed, as reports of weak signals obtained by individually underpowered studies accumulate in the literature, and particularly in view of the failure of standard statistical methods to appropriately indicate evidence against linkage, the overall picture of the schizophrenia genome becomes murkier, not clearer, as time goes on.
By design, repositories such as the HGI offer a solution by permitting analysis of far larger quantities of data than can be collected by any one study. However, under exactly these same conditions of underlying genetic complexity, the so-called mega-analyses, which simply pool all data into a single file for analysis, can also end up washing out important subset-specific loci by failing to appropriately allow for heterogeneity between data sets. For these reasons, we view the PPL—which does not require agreement across all data subsets, but rather accumulates the aggregate evidence both for and against linkage in a mathematically rigorous manner while allowing for differences between data sets—as particularly well suited to the task of accurately and efficiently extracting genetic information from repository collections. With the advent of affordable sequence data, we predict that revisiting family data already amassed in the HGI will provide a cost-effective mechanism not just for discovering linkage peaks, but for fine-mapping these peaks down to the level of the individual gene or variant.
1 : KELVIN: a software package for rigorous measurement of statistical evidence in human genetics. Hum Hered 2011; 72:276–288Crossref, Medline, Google Scholar
2 : NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet 1998; 81:282–289Crossref, Medline, Google Scholar
3 : Genome scan of European-American schizophrenia pedigrees: results of the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet 1998; 81:290–295Crossref, Medline, Google Scholar
4 : Genomewide linkage scan of 409 European-ancestry and African American families with schizophrenia: suggestive evidence of linkage at 8p23.3-p21.2 and 11p13.1-q14.1 in the combined sample. Am J Hum Genet 2006; 78:315–333Crossref, Medline, Google Scholar
5 : Genome scan of Han Chinese schizophrenia families from Taiwan: confirmation of linkage to 10q22.3. Am J Psychiatry 2006; 163:1760–1766Link, Google Scholar
6 : A genome screen for quantitative trait loci influencing schizophrenia and neurocognitive phenotypes. Am J Psychiatry 2008; 165:1185–1192Link, Google Scholar
7 : A genome-wide scan for schizophrenia and psychosis susceptibility loci in families of Mexican and Central American ancestry. Am J Med Genet B Neuropsychiatr Genet 2007; 144B:193–199Crossref, Medline, Google Scholar
8 : A schizophrenia gene locus on chromosome 17q21 in a new set of families of Mexican and Central American ancestry: evidence from the NIMH Genetics of Schizophrenia in Latino Populations Study. Am J Psychiatry 2009; 166:442–449Link, Google Scholar
9 : Linkage analysis of schizophrenia in African-American families. Schizophr Res 2009; 109:70–79Crossref, Medline, Google Scholar
10 : Practical considerations for dividing data into subsets prior to PPL analysis. Hum Hered 2008; 66:223–237Crossref, Medline, Google Scholar
11 : A multilocus model of the genetic architecture of autoimmune thyroid disorder, with clinical implications. Am J Hum Genet 2008; 82:1349–1356Crossref, Medline, Google Scholar
12 ;
13 : Identification of a schizophrenia-associated functional noncoding variant in NOS1AP. Am J Psychiatry 2009; 166:434–441Link, Google Scholar
14 : Testing for heterogeneity of recombination fraction values in human genetics. Ann Hum Genet 1963; 27:175–182Crossref, Medline, Google Scholar
15 : Power to detect linkage based on multiple sets of data in the presence of locus heterogeneity: comparative evaluation of model-based linkage methods for affected sib pair data. Hum Hered 2001; 51:199–208Crossref, Medline, Google Scholar
16 : Comparison of “model-free” and “model-based” linkage statistics in the presence of locus heterogeneity: single data set and multiple data set applications. Hum Hered 2001; 51:217–225Crossref, Medline, Google Scholar
17 : Simulated data for a complex genetic trait (problem 2 for GAW11): how the model was developed, and why. Genet Epidemiol 1999; 17(suppl 1):S449–S459Crossref, Medline, Google Scholar
18 : Association statistics under the PPL framework. Genet Epidemiol 2010; 34:835–845Crossref, Medline, Google Scholar
19 : The prior probability of autosomal linkage. Ann Hum Genet 1975; 38:341–350Crossref, Medline, Google Scholar
20 : Statistical Evidence: A Likelihood Paradigm. London, Chapman & Hall, 1997Google Scholar
21 : Review of “Statistical Evidence: A Likelihood Paradigm.” Am J Hum Genet 1998; 63:283–289Google Scholar
22 : Thermometers: something for statistical geneticists to think about. Hum Hered 2006; 61:144–156Crossref, Medline, Google Scholar
23 : Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet 2001; 10:3037–3048Crossref, Medline, Google Scholar
24 : Genomewide linkage scan of schizophrenia in a large multicenter pedigree sample using single nucleotide polymorphisms. Mol Psychiatry 2009; 14:786–795Crossref, Medline, Google Scholar
25 : A systematic genomewide linkage study in 353 sib pairs with schizophrenia. Am J Hum Genet 2003; 73:1355–1367Crossref, Medline, Google Scholar
26 : Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001; 98:4746–4751Crossref, Medline, Google Scholar
27 : Linkage analysis of a completely ascertained sample of familial schizophrenics and bipolars from Palau, Micronesia. Hum Genet 2005; 117:349–356Crossref, Medline, Google Scholar
28 : Genome-wide linkage scan of schizophrenia: a cross-isolate study. Genomics 2007; 89:167–177Crossref, Medline, Google Scholar
29 : Genome scan of three quantitative traits in schizophrenia pedigrees. Biol Psychiatry 2002; 52:847–854Crossref, Medline, Google Scholar
30 : Polymorphisms in SLC6A4, PAH, GABRB3, and MAOB and modification of psychotic disorder features. Schizophr Res 2009; 109:94–97Crossref, Medline, Google Scholar
31 : Aromatic amino acid hydroxylase genes and schizophrenia. Am J Med Genet 2002; 114:626–630Crossref, Medline, Google Scholar
32 : Phenylalanine hydroxylase gene in psychiatric patients: screening and functional assay of mutations. Biol Psychiatry 2003; 53:543–553Crossref, Medline, Google Scholar
33 : Convergent patterns of association between phenylalanine hydroxylase variants and schizophrenia in four independent samples. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:560–569Crossref, Medline, Google Scholar
34 : Common variants conferring risk of schizophrenia. Nature 2009; 460:744–747Crossref, Medline, Google Scholar
35 : Genome-wide search for schizophrenia susceptibility loci: the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet 1998; 81:275–281Crossref, Medline, Google Scholar
36 : A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr Res 2010; 124:192–199Crossref, Medline, Google Scholar
37 : Cross-disorder genomewide analysis of schizophrenia, bipolar disorder, and depression. Am J Psychiatry 2010; 167:1254–1263Link, Google Scholar
38 : Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet 2004; 74:886–897Crossref, Medline, Google Scholar
39 : Progress and pitfalls: bipolar molecular linkage studies. J Affect Disord 1998; 50:287–297Crossref, Medline, Google Scholar
40 : Genome scan in sibling pairs with juvenile-onset mood disorders: Evidence for linkage to 13q and Xq. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:638–646Crossref, Medline, Google Scholar
41 Nato A, Kong X, Byrne B, Naus J, Gordon D, Buyske S, Brzustowicz L, Matise T: Genomic characterization of schizophrenia candidate gene regions. American Society of Human Genetics Annual Meeting, Montreal, Oct 11–15, 2011. http://www.ichg2011.org/cgi-bin/showdetail.pl?absno=21931Google Scholar
42 : Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol Psychiatry 2009; 14:774–785Crossref, Medline, Google Scholar
43 : What association analysis can and cannot tell us about the genetics of complex disease. Am J Med Genet 1994; 54:318–323Crossref, Medline, Google Scholar
44 : The replication requirement. Nat Genet 2001; 29:244–245Crossref, Medline, Google Scholar
45 : Effect of allelic heterogeneity on the power of the transmission disequilibrium test. Genet Epidemiol 2000; 18:143–156Crossref, Medline, Google Scholar
46 : Problems of replicating linkage claims in psychiatry, in Genetic Approaches in Mental Disorders. Edited by Gershon ESCloninger CR. Washington, DC, American Psychiatric Press, 1994, pp 23–46Google Scholar

