
Psychobiological Mechanisms of Resilience and Vulnerability: Implications for Successful Adaptation to Extreme Stress
Abstract
OBJECTIVE: Most research on the effects of severe psychological stress has focused on stress-related psychopathology. Here, the author develops psychobiological models of resilience to extreme stress. METHOD: An integrative model of resilience and vulnerability that encompasses the neurochemical response patterns to acute stress and the neural mechanisms mediating reward, fear conditioning and extinction, and social behavior is proposed. RESULTS: Eleven possible neurochemical, neuropeptide, and hormonal mediators of the psychobiological response to extreme stress were identified and related to resilience or vulnerability. The neural mechanisms of reward and motivation (hedonia, optimism, and learned helpfulness), fear responsiveness (effective behaviors despite fear), and adaptive social behavior (altruism, bonding, and teamwork) were found to be relevant to the character traits associated with resilience. CONCLUSIONS: The opportunity now exists to bring to bear the full power of advances in our understanding of the neurobiological basis of behavior to facilitate the discoveries needed to predict, prevent, and treat stress-related psychopathology.
The adaptive physiological response to acute stress involves a process, initially referred to as allostasis by Sterling and Eyer (1), in which the internal milieu varies to meet perceived and anticipated demand. McEwen (2) extended this definition to include the concept of a set point that changes because of the process of maintaining homeostasis (2). The responses to severe stress that promote survival in the context of a life-threatening situation may be adaptive in the short run. However, if recovery from the acute event is not accompanied by an adequate homeostatic response to terminate the acute adaptive response of stress mediators, the deleterious effects on psychological and physiological function, termed the “allostatic load,” occur. The allostatic load is the burden borne by a brain and body adapting to challenges, both physiological and psychological. The concepts of allostasis and allostatic load link the protective and survival values of the acute response to stress to the adverse consequences that result if the acute response persists (3).
Much of the research on allostasis and allostatic load has focused on the negative effects of physiological stress on the brain and body. The present discussion will consider allostasis and allostatic load from the perspective of the effects of extreme psychological stress on the complex regulation of emotion by the brain and the consequences of such changes on human psychological resilience on one hand, and vulnerability to psychopathology on the other. Most of the neurobiological research on the consequences of severe psychological stress has focused on psychopathological responses that relate to stress-related disorders, such as posttraumatic stress disorder (PTSD) and major depression. Surprisingly, there has been little attention directed toward the question of which neurobiological responses are related to resilience to psychological stress in general and to specific forms of psychopathology.
Identification of responses that relate to psychobiological allostasis and reduced psychobiological allostatic load may provide clues toward discovering improved methods to prevent and treat disorders such as PTSD and major depression. For example, which aspects of the acute neurochemical response to traumatic stress promote behaviors that facilitate an effective survival reaction and may account for instances of highly effective action while experiencing fear? What psychobiological responses serve to maintain neural systems regulating reward and motivation in the face of an unrewarding environment? What alterations in neural systems regulating fear conditioning and extinction serve to maintain low levels of anxiety, despite an uncontrollable stress environment? Which changes in the neural systems involved in learning and memory can affect the encoding, consolidation, reconsolidation, and retrieval of memories of trauma so that normal psychological function can be maintained and re-experiencing symptoms minimized? How can neural systems regulating social behavior respond to persistent abuse and neglect to avoid a sense of hopelessness and interpersonal withdrawal? The answers to such questions may provide a greater understanding of why some individuals are able to cope with extreme stress with minimal psychopathological consequences.
A number of neurotransmitters, neuropeptides, and hormones have been linked to the acute psychobiological response to stress and the long-term psychiatric outcome. The roles of those neurotransmitters, neuropeptides, and hormones that have been shown to be significantly altered by psychological stress, have important functional interactions, and mediate the neural mechanisms and neural circuits relevant to the regulation of reward, fear conditioning, and social behavior will be reviewed. An attempt will be made to identify a putative neurochemical profile that characterizes psychobiological resilience and has predictive value regarding successful adaptation to extreme stress.
Cortisol and Dehydroepiandrosterone
There is consistent evidence that many forms of psychological stress increase the synthesis and release of cortisol. Cortisol serves to mobilize and replenish energy stores; it contributes to increased arousal, vigilance, focused attention, and memory formation; inhibition of the growth and reproductive system; and containment of the immune response. Cortisol has important regulatory effects on the hippocampus, amygdala, and prefrontal cortex (4). Glucocorticoids can enhance amygdala activity, increase corticotropin-releasing hormone (CRH) mRNA concentrations in the central nucleus of the amygdala (5–7), increase the effects of CRH on conditioned fear (8), and facilitate the encoding of emotion-related memory (9). Adrenal steroids such as cortisol have biphasic effects on hippocampal excitability and cognitive function and memory (10). These effects may contribute to adaptive alterations in behaviors induced by cortisol during the acute response to stress.
It is key, however, that the stress-induced increase in cortisol ultimately be constrained through an elaborate negative feedback system involving glucocorticoid and mineral corticoid receptors. Excessive and sustained cortisol secretion can have serious adverse effects, including hypertension, osteoporosis, immunosuppression, insulin resistance, dyslipidemia, dyscoagulation, and, ultimately, atherosclerosis and cardiovascular disease (11).
Another adrenal steroid released under stress is dehydroepiandrosterone (DHEA). DHEA is secreted episodically and synchronously with cortisol in response to fluctuating ACTH levels (12). DHEA has been shown to have antiglucocorticoid and antiglutamatergic activity in several tissues, including the brain (13), mediated by complicated mechanisms distinct from classical glucocorticoid receptor antagonism. Peripherally produced DHEA is thought to be a major source of brain DHEA. Within the brain, regionally specific metabolism of DHEA may ultimately control the nature of DHEA’s effects on cognition and behavior (14). For instance, 7-hydroxylated metabolites of DHEA in the hippocampus interfere with the normal uptake of activated glucocorticoid receptors (15) and may confer neuroprotection (16, 17). DHEA also restores cortisol-induced suppression of long-term potentiation in hippocampal neurons (18).
A negative correlation between DHEA reactivity to adrenal activation and the severity of PTSD has been reported, suggesting that enhanced DHEA release in response to prolonged stress may be protective in persons with PTSD (unpublished work by Rasmusson et al.). This is consistent with recent observations in a study of elite special operations soldiers that revealed negative correlations between ratios of DHEA to cortisol and dissociation during prolonged and extreme training stress and between DHEA and DHEA-S (“S” stands for sulfate) levels in the recovery period and better overall performance (unpublished work by Morgan et al.). Other evidence that suggests that DHEA promotes psychological resilience includes several studies reporting negative associations between plasma DHEA levels and depressive symptoms and the antidepressant effects of DHEA (19–22). Aside from the antiglucocorticoid actions of DHEA, effects on γ-aminobutyric acid (GABA)A receptors (23) and N-methyl-d-aspartic acid (NMDA)-based neurotransmission (24) may be involved in the behavioral effects of DHEA.
CRH
CRH is one of the most important mediators of the stress response, coordinating the adaptive behavioral and physiological changes that occur during stress (25). Release of CRH from the hypothalamus into the hypothalamic-pituitary portal circulation occurs in response to stress, resulting in activation of the hypothalamic-pituitary-adrenal (HPA) axis and the increased release of cortisol and DHEA. Equally important are the extrahypothalamic effects of CRH. CRH-containing neurons are located throughout the brain, including the prefrontal and cingulate cortices, the central nucleus of the amygdala, the bed nucleus of the stria terminalis, the nucleus accumbens, the periaqueductal gray matter, and the brainstem nuclei, such as the major norepinephrine-containing nucleus, the locus coeruleus, and the serotonin (5-HT) nuclei in the dorsal and median raphe (26).
Increased activity of amygdala CRH neurons activates fear-related behaviors, while cortical CRH may reduce reward expectation. CRH also inhibits a variety of neurovegetative functions, such as food intake, sexual activity, and endocrine programs for growth and reproduction. It appears that early-life stress can produce long-term elevation of brain CRH activity and that individual response to heightened CRH function may depend upon the social environment, past trauma history, and behavioral dominance (27). Persistent elevation of hypothalamic and extrahypothalamic CRH contributes mightily to the psychobiological allostatic load. Increased CSF levels of CRH have been linked to PTSD and major depression (28–30). Psychobiological resilience may be related to an ability to restrain the initial CRH response to acute stress.
Both CRH-1 and CRH-2 receptors are found in the pituitary and throughout the neocortex (especially in the prefrontal, cingulate, striate, and insular cortices), the amygdala, and the hippocampal formation in the primate brain. The presence of CRH-1 (but not CRH-2) receptors within the locus coeruleus, the nucleus of the solitary tract, the thalamus, the striatum, CRH-2 (but not CRH-1) receptors in the choroid plexus, certain hypothalamic nuclei, the nucleus prepositus, and the bed nucleus of the stria terminalis suggests that each receptor subtype has distinct roles within the primate brain (31).
CRH-1-deficient mice display decreased anxiety-like behavior and an impaired stress response (32). In contrast, CRH-2-deficient mice display increased anxiety-like behavior and are hypersensitive to stress (33, 34). Thus, evidence exists in favor of opposite functional roles for the two known CRH receptors; activation of CRH-1 receptors may be responsible for increased anxiety-like responses, and stimulation of CRH-2 may produce anxiolytic-like responses. Regulation of the relative contribution of the two CRH receptor subtypes to brain CSF pathways may be essential to coordinating psychological and physiological responses to stressors (32). Thus far, it has not been possible to evaluate CRH-1 and CRH-2 receptors in living human subjects, although efforts are ongoing to develop CRH receptor positron emission tomography ligands.
Locus Coeruleus-Norepinephrine System
Stress activates the locus coeruleus, which results in increased norepinephrine release in projection sites of the locus coeruleus, including the amygdala, the prefrontal cortex, and the hippocampus. The locus coeruleus is activated by a variety of stressors, both intrinsic (hypoglycemia, decreased blood volume, decreased blood pressure, altered thermoregulation, and distention of the colon and bladder) and extrinsic (environmental stress or threat) to the animal. Such activation is adaptive to survival from a life-threatening situation and serves as a general alarm function. Activation of the locus coeruleus also contributes to the sympathetic nervous system and HPA axis stimulation. Coincidentally, activation of the locus coeruleus inhibits parasympathetic outflow and neurovegetative function, including eating and sleep. A high level of activation of the locus coeruleus-norepinephrine system inhibits function of the prefrontal cortex, thereby favoring instinctual responses over more complex cognition (35).
The ability of acute stress to coactivate the HPA and locus coeruleus-norepinephrine systems facilitates the encoding and relay of aversively charged emotional memories, beginning at the amygdala. The amygdala also inhibits the prefrontal cortex (such as the locus coeruleus) and stimulates hypothalamic CRH release and brainstem autonomic centers, resulting in increased activity of the HPA and locus coeruleus. These feedback loops among the prefrontal cortex, amygdala, hypothalamus, and brainstem noradrenergic neurons contain the elements for a sustained and powerful stress response (4). If unchecked, persistent hyperresponsiveness of the locus coeruleus-norepinephrine system will contribute to chronic anxiety, fear, intrusive memories, and an increased risk of hypertension and cardiovascular disease. In some patients with panic disorder, PTSD, and major depression, there is evidence of heightened locus coeruleus-norepinephrine activity (36–40).
Neuropeptide Y
Neuropeptide Y is a highly conserved 36 amino acid peptide, which is among the most abundant peptides found in the mammalian brain. There are five brain areas in which neurons containing neuropeptide Y are densely concentrated: the locus coeruleus (41), the paraventricular nucleus of the hypothalamus (42), septohippocampal neurons (43), the nucleus of the solitary tract, and the ventral lateral medulla (44). Moderate levels are found in the amygdala, hippocampus, cerebral cortex, basal ganglia, and thalamus (45).
Evidence suggesting the involvement of the amygdala in the anxiolytic effects of neuropeptide Y is robust and probably occurs by means of the neuropeptide Y-Y1 receptor (46–48). Microinjection of neuropeptide Y into the central nucleus of the amygdala reduces anxious behaviors. The up-regulation of amygdala neuropeptide Y mRNA levels after chronic stress suggests that neuropeptide Y may be involved in the adaptive responses to stress exposure (49). Neuropeptide Y may also be involved in the consolidation of fear memories; injection of neuropeptide Y into the amygdala impairs memory retention in a foot-shock avoidance paradigm (50). The anxiolytic effects of neuropeptide Y also involve the locus coeruleus, possibly by means of the neuropeptide Y-Y2 receptor. Neuropeptide Y reduces the firing of neurons in the locus coeruleus (51). Neuropeptide Y also has behaviorally relevant effects on the hippocampus. Transgenic rats with hippocampal neuropeptide Y overexpression have attenuated sensitivity to the behavioral consequences of stress and impaired spatial learning (52).
There are important functional interactions between neuropeptide Y and CRH (53, 54). Neuropeptide Y counteracts the anxiogenic effects of CRH, and a CRH antagonist blocks the anxiogenic effects of a neuropeptide Y-Y1 antagonist (55). Thus, it has been suggested that the balance between neuropeptide Y and CRH neurotransmission is important to the emotional responses to stress (54). In general, brain regions that express CRH and CRH receptors also contain neuropeptide Y and neuropeptide Y receptors, and the functional effects are often opposite (56), especially at the level of the locus coeruleus (57, 58), amygdala (59, 60), and the periaqueductal gray matter (61, 62).
These data suggest an important role for an up-regulated neuropeptide Y system in the psychobiology of resilience. Neuropeptide Y has counterregulatory effects on both the CRH and locus coeruleus-norepinephrine systems at brain sites that are important in the expression of anxiety, fear, and depression. Preliminary studies in special operations soldiers under extreme training stress indicate that high neuropeptide Y levels are associated with better performance (63). Patients with PTSD have been shown to have reduced plasma neuropeptide Y levels and a blunted yohimbine-induced neuropeptide Y increase (64). Additionally, low levels of neuropeptide Y have been found in depressed patients, and a variety of antidepressant drugs increase neuropeptide Y levels (65).
Galanin
Galanin is a peptide that, in humans, contains 30 amino acids. It has been demonstrated to be involved in a number of physiological and behavioral functions, including learning and memory, pain control, food intake, neuroendocrine control, cardiovascular regulation, and, most recently, anxiety (66).
Galanin is closely associated with ascending monoamine pathways. Approximately 80% of noradrenergic cells in the locus coeruleus co-express galanin. A dense galanin immunoreactive fiber system originating in the locus coeruleus innervates forebrain and midbrain structures, including the hippocampus, hypothalamus, amygdala, and prefrontal cortex (67–69). Neurophysiological studies have shown that galanin reduces the firing rate of the locus coeruleus, possibly by stimulating the galanin-1 receptor, which acts as an autoreceptor (70, 71).
Studies in rats have shown that galanin administered centrally modulates anxiety-related behaviors (72, 73). Galanin-overexpressing transgenic mice do not exhibit an anxiety-like phenotype when tested under baseline (nonchallenged) conditions. However, these mice are unresponsive to the anxiogenic effects of the alpha-2 receptor antagonist yohimbine. Consistent with this observation, galanin administered directly into the central nucleus of the amygdala blocked the anxiogenic effects of stress, which is associated with increased norepinephrine release in the central nucleus of the amygdala. Yohimbine increases galanin release in the central nucleus of the amygdala (74). Galanin administration and galanin overexpression in the hippocampus result in deficits in fear conditioning (75).
The mechanism by which galanin reduces norepinephrine release at locus coeruleus projections to the amygdala, hypothalamus, and prefrontal cortex may be a direct action of galanin on these brain regions by means of galanin-synthesizing neurons or by stimulating galanin receptors in these regions (71, 74). It is not known which galanin receptors are involved. Galanin-1 receptor mRNA levels are high in the amygdala, hypothalamus, and bed nucleus of the stria terminalis (76), and galanin-1 receptor-deficient mice show increased anxiety-like behavior (77).
These results suggest that the noradrenergic response to stress can recruit the release of galanin in the central nucleus of the amygdala and prefrontal cortex, which then buffers the anxiogenic effects of norepinephrine. Thus, the net behavioral response due to stress-induced noradrenergic hyperactivity may depend upon the balance between norepinephrine and neuropeptide Y and galanin neurotransmission. This hypothesis is consistent with evidence that release of neuropeptides preferentially occurs under conditions of high neurotransmitter activity (78, 79). To our knowledge, galanin function has not been studied in patients exposed to traumatic stress or patients with PTSD or major depression. Galanin and neuropeptide Y receptor agonists may be novel targets for the development of antianxiety drugs (71).
Dopamine
Uncontrollable stress activates dopamine release in the medial prefrontal cortex (80) and inhibits dopamine release in the nucleus accumbens (81, 82). Lesions of the pretraining and posttraining amygdala in a conditioned stress model block stress-induced dopamine metabolic activation in the medial prefrontal cortex, suggesting amygdala control of stress-induced dopamine activation and a role for integrating the behavioral and neuroendocrine components of the stress response (83). There is preclinical evidence that the susceptibility of the mesocortical dopamine system to stress activation may be in part genetically determined. It has been suggested that excessive mesocortical dopamine release by stressful events may represent a vulnerability to depression and favor helpless reactions through an inhibition of subcortical dopamine transmission (80, 82). These observations may be due to the effect of dopamine on reward mechanisms.
On the other hand, lesions of dopamine neurons in the medial prefrontal cortex delay extinction of the conditioned fear stress response (no effect on acquisition), indicating that prefrontal dopamine neurons are involved in facilitating extinction of the fear response. This suggests that reduced prefrontal cortical dopamine results in the preservation of fear produced by a conditioned stressor, a situation hypothesized to occur in PTSD (84). One way to reconcile these two sets of data is to suggest that there is an optimal range for stress-induced increases in cortical dopamine released in the medial prefrontal cortex to facilitate adaptive behavioral responses. Too much dopamine release in the medial prefrontal cortex produces cognitive impairment; an inhibition in dopamine activity in the nucleus accumbens results in abnormalities in motivation and reward mechanisms. Insufficient prefrontal cortical dopamine release delays extinction of conditioned fear. There has been little clinical research regarding dopamine function as it pertains to stress-related psychopathology. Several clinical investigations have reported increased urinary and plasma dopamine concentrations (85, 86) in PTSD. In contrast, reduced dopamine metabolism has been demonstrated in depressed patients (87).
Serotonin
Different types of acute stress result in increased 5-HT turnover in the prefrontal cortex, nucleus accumbens, amygdala, and lateral hypothalamus (88). Serotonin release may have both anxiogenic and anxiolytic effects, depending on the region of the forebrain involved and the receptor subtype activated. For example, anxiogenic effects are mediated by means of the 5-HT2A receptor, whereas stimulation of 5-HT1A receptors are anxiolytic and may even relate to adaptive responses to aversive events (89).
Understanding the function of the 5-HT1A receptor is probably most pertinent to the current discussion. The 5-HT1A receptors are found in the superficial cortical layers, the hippocampus, the amygdala, and the raphe nucleus (primarily presynaptic) (90, 91). The behavioral phenotype of 5-HT1A knockout mice includes increases in anxiety-like behaviors (92, 93). These behaviors are mediated by postsynaptic 5-HT1A receptors in the hippocampus, amygdala, and cortex (94). Of great interest is the recent finding that embryonic and early postnatal shutdown of expression of 5-HT1A receptors produces an anxiety phenotype that cannot be rescued with restoration of 5-HT1A receptors. However, when 5-HT1A receptor expression is reduced in adulthood and then reinstated, the anxiety phenotype is no longer present. These results suggest that altered function of 5-HT1A receptors early in life can produce long-term abnormalities in the regulation of anxiety behaviors (94).
Postsynaptic 5-HT1A receptor gene expression is under tonic inhibition by adrenal steroids such as in the hippocampus, apparently mostly by means of activation of mineral corticoid receptors. 5-HT1A receptor density and mRNA levels decrease in response to stress, which is prevented by adrenalectomy (95).
There may also be important functional interactions between 5-HT1A and benzodiazepine receptors. In one study of 5-HT1A knockout mice, a down-regulation of benzodiazepine GABA α1 and α2 receptor subunits, as well as benzodiazepine-resistant anxiety in the elevated-plus maze was reported (96). However, a subsequent study did not replicate these results using mice with a different genetic background (97), raising the possibility that genetic background can affect functional interplay between 5-HT1A and benzodiazepine systems.
These results suggest a scenario in which early-life stress increases CRH and cortisol levels, which, in turn, down-regulate 5-HT1A receptors, resulting in a lower threshold for anxiogenic stressful life events. Alternatively, 5-HT1A receptors may be decreased on a genetic basis. The density of 5-HT1A receptors is reduced in depressed patients when they are depressed as well as in remission (98). It has been recently demonstrated that 5-HT1A receptor density is also decreased in patients with panic disorder (99). Examination of 5-HT1A receptor density in patients with anxiety disorders is indicated.
Benzodiazepine Receptors
Animals exposed to chronic inescapable stress develop behaviors that are consistent with excessive fear and anxiety, such as increased fearfulness, increased defecation, and avoidance of novel situations (e.g., an open field). Exposure to inescapable stressors produce decreases in benzodiazepine receptor binding in the cortex, with some studies showing a decrease in the hippocampus (100, 101). Exposure to stress has no effects on benzodiazepine receptor binding in the pons, striatum, thalamus, cerebellum, midbrain, or occipital cortex. These data support a role for alterations in benzodiazepine binding in anxiety, with a specific decrease in the frontal cortex and, although not as consistently, a decrease in the hippocampus (101).
Neuroimaging studies reveal reduced cortical and subcortical benzodiazepine receptor binding in patients with PTSD and panic disorder (102–104). The findings could be related to a down-regulation of benzodiazepine receptor binding after exposure to stress. Other possible explanations are that stress results in changes in receptor affinity, changes in an endogenous benzodiazepine ligand (the existence of which is controversial), and stress-related alterations in GABAergic transmission or neurosteroids that affect benzodiazepine receptor binding. A preexisting low level of benzodiazepine receptor density may be a genetic risk factor for the development of stress-related anxiety disorders.
Gonadal Steroids
Testosterone
Testosterone has been among the most studied of all hormones in terms of its relationships to specific behaviors. Aggression is the aspect of human behavior most often linked to testosterone concentrations (105). Preclinical studies consistently show that low levels of testosterone are associated with submissive behavior. In mandrils and squirrel monkeys, social rank correlates with testosterone levels (105, 106). In human subjects, the personal experience of success, as well as the feeling of dominance in a competitive situation, is associated with higher testosterone levels (107, 108). Increased levels of testosterone have been found in male prison inmates with frequent episodes of violent behavior (109–111). Psychological stress is associated with decreases in testosterone levels. For example, elite soldiers participating in a physically and psychologically stressful training exercise show a lowering of their testosterone levels (63).
The mechanism by which testosterone is reduced by physical and psychological stress remains to be elucidated. It is unclear whether the decrease in testosterone from exposure to mental stress is caused by decreased leuteinizing hormone-releasing hormone (LH-RH) synthesis at the hypothalamus or leuteinizing hormone (LH) secretion in the pituitary (105). Perhaps a more likely mechanism involves a recently identified hypothalamic-testicular pathway that is independent of the pituitary but travels through the spinal cord. This pathway appears to mediate the effect of CRH to decrease testosterone levels. Thus, hypothalamic increases in CRH produced by psychological stress may be associated with decreased testosterone by stimulating the neural pathway that interferes with Leydig cell function independently of the pituitary. It is important to establish the relative role of the LH-RH/LH axis and the hypothalamic testicular axis in modulating the influence of specific stressors on testosterone release (112).
There is a recent report of reduced CSF testosterone levels in PTSD patients that was negatively correlated with CSF CRH concentrations (113). There was no correlation between plasma and CSF testosterone levels (113). The data from studies measuring plasma testosterone levels in PTSD patients are mixed (114).
Depressed men have been found to have decreased serum or plasma testosterone in some studies (115), but not all, because of confounding factors. Hypogonadal men often experience depressive symptoms, which are improved by testosterone-replacement therapy (116). Clinical trials of depressed men with decreased testosterone have produced contradictory results. However, a recent placebo-controlled study (115) found testosterone gel to be effective for men with treatment-resistant depression and low testosterone levels when added to an existing antidepressant regimen. Testosterone administration may be helpful for patients with low testosterone secondary to chronic severe psychological stress.
Estrogen
There is abundant preclinical and clinical literature demonstrating consistent gender differences in stress responsiveness (117). Most of the work focused on HPA responses to stressors. Female rats consistently show greater increases in corticosterone and ACTH in response to acute and chronic stressors. These differences have generally been attributed to the activational effects of gonadal steroids on elements of the HPA axis in females (118). Several studies suggest that estradiol plays a role in enhanced stress responses in female rats, based upon increased HPA axis responses to stress when ovariectomized rats are treated with estradiol (119). A possible mechanism for these findings is that estrogen (as well as progesterone) produces a relative resistance to glucocorticoid feedback (120).
However, a recent investigation by Young and colleagues, studying the effects of estrogen antagonists and physiological doses of estradiol, found that estradiol reduced the ACTH response to restraint stress in female rats (118). The estrogen antagonists had the opposite effect. These data suggest that physiological doses of estradiol are inhibitory to stress responsiveness and that blocking estradiol on gonadally intact, normally cycling female rats leads to exaggerated stress responsiveness. The contrast with prior studies seems to relate to the dosage of estradiol and the duration of administration. Considered together, the studies indicate that short-term exposure to low doses of estrogen can suppress HPA axis responses to stress but higher doses and more prolonged treatment enhances HPA axis responses (117, 118). The mechanism underlying these effects could be due to enhanced negative feedback or decreases in the stimulatory aspects of the system, related to either CRH or ACTH. This remains to be elucidated, since studies examining the effects of estradiol on mineral corticoid receptor and glucocorticoid receptor binding and mRNA expression and on CRH have not yielded consistent results, perhaps due to variability in doses and duration of treatment regimens.
Studies in human populations suggest that female subjects respond with greater HPA activation to stressors involving interpersonal concerns (social rejection) and male subjects to achievement-oriented stressors (117). The role of estrogen in these differential responses remains to be studied. Estrogen has been shown to blunt HPA axis responses to psychological stress in postmenopausal women (121, 122) and to blunt the ACTH response to CRH in postmenopausal women with high levels of body fat. In addition, 8 weeks of estrogen supplementation to perimenopausal women blunted systolic and diastolic blood pressure, cortisol, ACTH, plasma epinephrine and norepinephrine, and norepinephrine responses of the entire body to stress (120).
Although the mechanisms responsible for the effect of estrogen on glucocorticoid levels are not fully defined, it appears that it acts by means of ACTH and thus the pituitary or hypothalamus rather than directly on the adrenal gland. This is consistent with evidence obtained from women with hypothalamic amenorrhea, in whom a blunted response to CRH administration and increased cortisol levels were observed (123). These effects could be explained by a direct action of estrogen on CRH gene expression or glucocorticoid receptor numbers or function (124).
The mechanisms by which estrogens affect catecholamine levels are also uncertain. The effects of estrogen may be due to actions on the adrenal gland or central or peripheral neuronal pathways. Neuronal pathways seem more likely (125), although several different mechanisms may be involved, including effects on α1-noradrenergic (126) and β-noradrenergic (127) receptors and modulation of norepinephrine release. Estrogen has also been shown to up-regulate the GABAA benzodiazepine receptor (128).
The effects of estrogen on mood and anxiety may be mediated in part by the serotonin system (129). Estrogen has complex effects on functioning of the serotonin system, including increased tryptophan hydroxylase gene and protein expression (130), decreased expression of the serotonin transporter (131), and increased 5-HT2A binding (132). Perhaps most important are studies relevant to the 5-HT1A receptor. Estrogen in both rats and monkeys decreases 5-HT1A in RNA and 5-HT1A binding in both presynaptic (dorsal raphe) and postsynaptic sites (133). Estrogen also decreases the inhibitory G proteins involved in intracellular signal transduction mediated by the 5-HT1A receptor (134, 135).
Women appear to be more sensitive to the effects of traumatic stress. One survey found that 31% of women and 19% of men develop PTSD when exposed to major trauma (136). However, the role of estrogen in the development of PTSD has not been investigated. Based upon these data, short-term increases in estrogen after exposure to stress might be beneficial because of its ability to blunt the HPA axis and noradrenergic response to stress. However long-term stress-related elevation in estrogen might be detrimental because of estrogen-induced decreases in 5-HT1A receptor numbers and function.
Resilience and Vulnerability to Stress
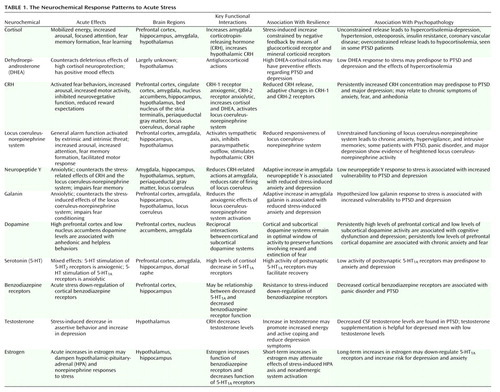
The last section identified 11 possible mediators of the psychobiological response to extreme stress and how each may contribute, alone or through functional interactions, to resilience or vulnerability (Table 1 and Figure 1). In the beginning of this article, the concept of allostatic load was introduced as a measure of the cumulative physiological burden borne by the body from attempts to adapt to stressors and strains of life’s demands (137). McEwen and Stellar (3) hypothesized that the cumulative impact on health risk from modest dysregulations in multiple systems can be substantial, even if they individually have minimal and insignificant health effects. Thus, they defined allostatic load as a cumulative measure of physiological dysregulation over multiple systems (3).
The concept of allostatic load has proven to be useful as a predictor of functional decline in elderly men and women. Seeman and colleagues (138) developed a measure of allostatic load based on 10 markers reflecting levels of physiological activity across a range of important regulatory systems, which individually have been linked to disease based upon data from a longitudinal community-based study of successful aging (138). The markers were the following:
| 1. | Twelve-hour overnight urinary cortisol excretion | ||||
| 2. | Twelve-hour overnight urinary excretion of norepinephrine | ||||
| 3. | Twelve-hour overnight urinary excretion of epinephrine | ||||
| 4. | Serum DHEA-S level | ||||
| 5. | Average systolic blood pressure | ||||
| 6. | Average diastolic blood pressure | ||||
| 7. | Ratio of waist-hip circumference | ||||
| 8. | Serum high-density lipid (HDL) cholesterol | ||||
| 9. | Ratio of total cholesterol to HDL cholesterol | ||||
| 10. | Blood-glycosylated hemoglobin | ||||
For each of the 10 markers, the subjects were classified into quartiles based upon the distribution of scores in the baseline cohort. Allostatic load was measured by summing the number of parameters for which the subject fell into the highest-risk quartile (top quartile for all markers except HDL cholesterol and DHEA-S, for which the lowest quartile corresponds to the highest risk). In two follow-up studies encompassing 2.5 and 7 years, none of the 10 markers of allostatic load exhibited significant associations on their own with health outcomes. However, the summaried measure of allostatic load was found to be significantly associated with four major health outcomes: 1) new cardiovascular events, 2) a decline in cognitive functioning, 3) a decline in physical functioning, and 4) mortality. Thus, these data are consistent with the hypothesis that although modest abnormalities in a single physiological system may not be predictive of poor health outcome, the cumulative effect of multiple abnormalities in the physiological system is prognostic of poor physical health (11, 138).
The allostatic load concept has not been used to investigate neurobiological risk factors related to psychopathology. Perhaps an analogous approach that involves the identification of a group of biological markers that will relate to psychobiological allostasis and psychobiological allostatic load and, consequently, to resilience and vulnerability to the effects of extreme psychobiological stress will be fruitful. It is in this context that this review of the neurochemical response patterns to stress can provide a framework for developing a measure for psychobiological allostatic load. The finding that many of these measures have important functional interactions is supportive of the concept of developing a more integrative measure. One prediction is that individuals in the highest quartile for measures of HPA axis, CRH, locus coeruleus-norepinephrine, dopamine, and estrogen activity and the lowest quartile for DHEA, neuropeptide Y, galanin, testosterone, and 5-HT1A receptor and benzodiazepine receptor function will have the highest index for psychobiological allostatic load and an increased risk for psychopathology after exposure to stress. It is possible that psychobiological allostatic load will relate to vulnerability to the effects of chronic, mild, intermittent stressors as well as extreme psychological trauma. In contrast, a resilient profile will be characterized by individuals in the highest quartile for measures of DHEA, neuropeptide Y, galanin, testosterone, and 5-HT1A receptor and benzodiazepine receptor function and the lowest quartile for HPA axis, CRH, and locus coeruleus-norepinephrine activity (Table 1). The mediators of the stress response identified in this review are not meant to be an exhaustive or definitive list. For example, glutamate and neurotrophic factors, such as brain-derived neurotrophic factor, and neuropeptides, such as substance P and cholecystokinin, could have been included. Longitudinal community-based surveys of successful adaptation to extreme stress should be considered to determine if markers such as these or others can be used to develop a measure of psychobiological allostatic load that will be of predictive value.
Reward, Fear Conditioning, and Social Behavior
Most of the research on resilience in the face of adversity focuses on early childhood and adolescence. Studies of children raised in a variety of settings, including war, family violence, poverty, and natural disasters, have revealed a consistent pattern of individual characteristics associated with successful adaptation. These include good intellectual functioning, effective self-regulation of emotions and attachment behaviors, a positive self-concept, optimism, altruism, a capacity to convert traumatic helplessness into learned helpfulness, and an active coping style in confronting a stressor (139–141).
Which adult characteristics are associated with resilience to stress? Most of the data come from studies of men in combat but are applicable to other professions, such as firefighters and police, in which danger is ever-present and effective action under stress is imperative. These include an ability to bond with a group with a common mission, a high value placed on altruism, and the capacity to tolerate high levels of fear and still perform effectively. Most courageous individuals are not fearless but are willing and able to approach a fear-inducing situation despite the presence of subjective fear and psychophysiological disturbance. For example, the original Mercury 7 astronauts reported that they had encountered challenges in which they felt fear but still were able to function effectively (142–144).
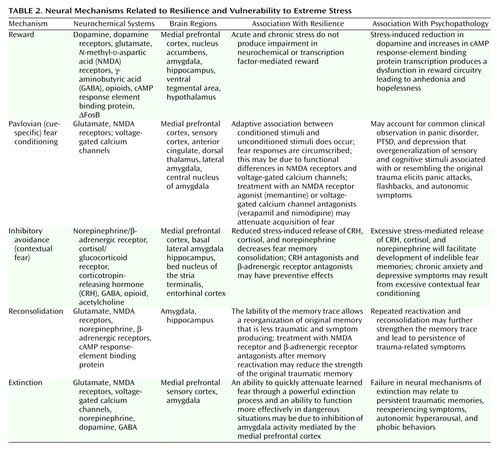
In recent years, significant advances have been made in understanding how the brain regulates reward and motivation (hedonia, optimism, and learned helpfulness), learns, remembers, and responds to fear (effective behaviors despite fear), and develops adaptive social behaviors (altruism, bonding, and teamwork). The neural mechanisms that mediate these functions are relevant to how an individual responds to extreme stress and may account, at least in part, for the character traits reviewed that relate to resilience and courage (Table 2).
Regulation of Reward
The ability to maintain properly functioning reward pathways and a hedonic tone in the context of chronic stress and an unrewarding environment may be critical to maintaining optimism, hopefulness, and a positive self-concept after exposure to extreme stress. Resilient individuals may have a reward system that is either hypersensitive to reward or is resistant to change, despite chronic exposure to neglect and abuse.
The mesolimbic dopamine pathways are centrally involved in reward, motivation, and hedonic tone. Subcortical structures involved in dopamine signaling include the dorsal striatum, ventral striatum (i.e., nucleus accumbens), amygdala, and midbrain ventral tegmental area (145, 146). The nucleus accumbens and its dopaminergic inputs play a central role in reward. The nucleus accumbens is a target of the mesolimbic dopamine system, which arises in the ventral tegmental area. The neurons of the ventral tegmental area also innervate several other limbic structures, including the amygdala and the medial prefrontal cortex. The amygdala sends projections to the ventral tegmental area and nucleus accumbens. Increasing evidence suggests that similar mechanisms in the ventral tegmental area and nucleus accumbens mediate responses to natural reinforcers under normal conditions. In nonhuman primates, the firing patterns of dopamine neurons in the ventral tegmental area are sensitive readouts of reward expectations. Dopamine neurons increase their firing relative to the predictability of reward. The dopamine neuronal response is activated when rewards occur without being predicted or are better than predicted. The neurons show no change when rewards are predicted and decreased activity when rewards are omitted or are less than predicted (147, 148).
The medial prefrontal cortex receives glutamatergic input from the amygdala and sends glutamatergic projections to the nucleus accumbens and the ventral tegmental area. Electrical stimulation of the medial prefrontal cortex is thought to be rewarding because it causes glutamate release in the ventral tegmental area and dopamine release in the nucleus accumbens. In contrast, the drug of abuse phencyclidine is rewarding because of its antagonism of NMDA-type glutamate receptors in the nucleus accumbens and the medial prefrontal cortex. Functional interactions among glutamate, NMDA receptors, dopamine, and dopamine receptors are critical to the proper functioning of reward circuits (146, 147, 149). Neurons of the orbital-frontal cortex, which receive dopamine projections from the ventral tegmental area, have the ability to discriminate different rewards according to their motivational value. The preference-related activations may facilitate neuronal mechanisms that lead to behavioral choices favoring the most rewarding and profitable goals (147, 148).
Recent approaches to reward mechanisms include examination of the molecular and cellular changes in the ventral tegmental area and nucleus accumbens pathway. Acute and chronic stress induce transcription in the nucleus accumbens that is mediated by cAMP response-element binding protein (150). This is associated with increased sensitivity to aversive stimuli and decreased sensitivity to rewarding stimuli. Thus, cAMP response-element binding protein in the nucleus accumbens modulates behavioral responsiveness to emotional stimuli such that increased cAMP response-element binding protein after stress may contribute to persistent anhedonia in patients with PTSD or major depression (145).
The amygdala modulates conditioned responses to rewarding stimuli through circuits formed by the amygdala, subiculum, bed nucleus of the stria terminalis, nucleus accumbens, and medial prefrontal cortex. These neural networks establish the emotional value of a reward memory as well as its strength and persistence. The molecular basis for such plasticity is just beginning to be developed—the cAMP pathway and cAMP response-element binding protein in the amygdala promote both aversive and rewarding associations (151, 152).
Sensitivity to the behavioral effects of dopamine-enhancing drugs may be heritable. There may be an endophenotype related to resistance to anhedonia and hopelessness in the face of stress (153). Subjects with major depression are hyperresponsive to amphetamine such that the severity of depression in major depression was highly correlated with the rewarding effects of amphetamine. The mechanism may be depletion of synaptic dopamine with up-regulation of dopamine receptors (154, 155). Increasing dopamine function in the nucleus accumbens, the orbital frontal cortex, and the ventral tegmental area and NMDA receptor blockade in the nucleus accumbens and the medial prefrontal cortex may enhance sensitivity to reward. Therefore, psychostimulants, dopamine reuptake inhibitors, monoxamine oxidase-B inhibitors (selegiline), the dopamine receptor agonists (pramipexole), and NMDA receptor antagonists (memantine) may be useful for treating anhedonia and hopelessness resulting from traumatic stress exposure.
The Neural Mechanisms of Anxiety and Fear
Fear Conditioning
Fear conditioning in many patients with PTSD and major depression causes vivid recall of memories of traumatic events, autonomic hyperarousal, and even flashbacks elicited by sensory and cognitive stimuli associated with prior traumas. Consequently, patients may begin to avoid these stimuli in their everyday lives, or a numbing of general emotional responsiveness may ensue. Resilience to the effects of severe stress may be characterized by the capacity to avoid overgeneralizing specific conditioned stimuli to a larger context, reversible storage of emotional memories, and facilitated extinction.
Classical fear conditioning is a form of associative learning in which subjects come to express fear responses to a neutral conditioned stimulus that is paired with an aversive unconditioned stimulus. The conditioned stimulus, as a consequence of this pairing, acquires the ability to elicit a spectrum of behavioral, autonomic, and endocrine responses that normally would only occur in the context of danger (156). Fear conditioning can be adaptive and enable efficient behavior in dangerous situations. The individual who can accurately predict threat can engage in appropriate behaviors in the face of danger. In the clinical situation, specific environmental features (conditioned stimuli) may be linked to the traumatic event (unconditioned stimuli), such that reexposure to a similar environment produces a recurrence of the symptoms of anxiety and fear. Patients often generalize these cues and experience a continuous perception of threat to the point that they become conditioned to context.
Cue-specific conditioned stimuli are transmitted to the thalamus by external and visceral pathways. Afferents then reach the lateral amygdala by means of two parallel circuits: a rapid subcortical path directly from the dorsal (sensory) thalamus and a slower regulatory cortical pathway encompassing the primary somatosensory cortices, the insula, and the anterior cingulate/prefrontal cortex. Contextual conditioned stimuli are projected to the lateral amygdala from the hippocampus and perhaps the bed nucleus of the stria terminalis. The long loop pathway indicates that sensory information relayed to the amygdala undergoes substantial higher-level processing, thereby enabling assignment of significance based on prior experience to complex stimuli. Cortical involvement in fear conditioning is clinically relevant because it provides a mechanism by which cognitive factors will influence whether symptoms are experienced or not following exposure to stress (157).
During the expression of fear-related behaviors, the lateral amygdala engages the central nucleus of the amygdala, which, as the principal output nucleus, projects to areas of the hypothalamus and brainstem that mediate the autonomic, endocrine, and behavioral responses associated with fear (158). The molecular and cellular mechanisms that underlie synaptic plasticity in amygdala-dependent learned fear are an area of active investigation (159). Long-term potentiation in the lateral amygdala appears to be a critical mechanism for storing memories of the association between conditioned stimuli and unconditioned stimuli (156). A variety of behavioral and electrophysiological data have led LeDoux and colleagues (157, 158) to propose a model to explain how neural responses to the conditioned stimuli and unconditioned stimuli in the lateral amygdala could influence long-term potentiation-like changes that store memories during fear conditioning. This model proposes that calcium entry through NMDA receptors and voltage-gated calcium channels initiates the molecular processes to consolidate synaptic changes into long-term memory (156). Short-term memory requires calcium entry only through NMDA receptors and not voltage-gated calcium channels.
This hypothesis leads to several predictions that may have relevance to psychological responses to stress. It suggests that blocking NMDA receptors in the amygdala during learning should impair memory of short- and long-term fear. This has been demonstrated in rodents (160, 161). Valid human models of fear conditioning and the availability of the NMDA receptor antagonist memantine should permit this hypothesis to be tested clinically (162). If memantine impairs the acquisition of fear in humans, it may have use in the prevention and treatment of stress-induced disorders such as PTSD. Blockade of voltage-gated calcium channels appears to block long-term but not short-term memory (163). Therefore, clinically available calcium channel blockers such as verapamil and nimodipine may be helpful in diminishing the intensity and impact of recently acquired fear memory and perhaps in preventing PTSD as well.
This discussion has focused primarily upon the neural mechanisms related to the coincident learning of the unconditioned stimuli-conditioned stimuli association (i.e., Pavlovian fear conditioning) in the lateral amygdala. However, there is significant evidence that a broader neural circuitry underlies the memory of fear that is modulated by amygdala activity. The inhibitory-avoidance paradigm is used to examine memory consolidation for aversively motivated tasks and involves intentional instrumental choice behavior. Studies using inhibitory avoidance learning procedures have been used to support the view that the amygdala is not the sole site for fear learning but, in addition, can modulate the strength of memory storage in other brain structures (164).
There is evidence that Pavlovian fear conditioning and inhibitory avoidance involve fundamentally different neural mechanisms. Pavlovian fear conditioning and inhibitory avoidance are differentially affected by posttraining pharmacological manipulations. The two types of learning involve different experimental procedures. In Pavlovian fear conditioning, the presentation of the conditioned stimuli and unconditioned stimuli occurs independent of behavior, whereas with inhibitory avoidance shock, delivery is contingent upon an animal’s behavioral response. Inhibitory avoidance may involve a more complex neural network because an animal’s response is contingent upon a number of contextual cues, in contrast to the more specific conditioned stimuli and unconditioned stimuli. The basal lateral amygdala is the primary amygdala nucleus responsible for voluntary emotional behavior based upon aversive emotional events, whereas the central nucleus of the amygdala is more involved in Pavlovian responses to fear-inducing stimuli (165). The relevance of the inhibitory-avoidance paradigm to human fear and anxiety rests on its assessment of a behavioral response to a fear-inducing context (166).
Specific drugs and neurotransmitters infused into the basal lateral amygdala influence consolidation of memory for inhibitory avoidance training. Posttraining peripheral or intra-amygdala infusions of drugs affecting GABA, opioid, glucocorticoid, and muscarinic acetylcholine receptors have dose- and time-dependent effects on memory consolidation (164). Norepinephrine infused directly into the basal lateral amygdala after inhibitory avoidance training enhances memory consolidation, indicating that the degree of activation of the noradrenergic system within the amygdala by an aversive experience may predict the extent of the long-term memory for the experience (167).
Interactions among CRH, cortisol, and norepinephrine receptors have important effects on memory consolidation, which is likely to be relevant to the effects of traumatic stress on memory. Extensive evidence indicates that glucocorticoids influence long-term memory consolidation by means of stimulation of glucocorticoid receptors. The glucocorticoid effects on memory consolidation require activation of the basal lateral amygdala, and lesions of the basal lateral amygdala block retention enhancement of intrahippocampal infusions of a glucocorticoid receptor agonist. Additionally, the basal lateral amygdala is a critical locus of interaction between glucocorticoids and norepinephrine in modulating memory consolidation (168).
There is also extensive evidence consistent with a role for CRH in mediating the effects of stress on memory consolidation. Activation of CRH receptors in the basal lateral amygdala by CRH released from the central nucleus of the amygdala facilitates the effects of stress on memory consolidation. As reviewed, there are important functional interactions between the CRH and norepinephrine systems, including a role in memory consolidation. Memory enhancement produced by CRH infusions in the hippocampus are blocked by propranolol and the noradrenergic toxin DSP-4 (75-R), suggesting that CRH infusions by means of a presynaptic mechanism stimulate norepinephrine release in the hippocampus (169).
Efferent projections from the basal lateral amygdala are also crucial to memory formation. The basal lateral pathway of the amygdala stria terminalis is involved, since lesions of the stria terminalis impair the memory-enhancing effects of intra-amygdala infusions of norepinephrine and systemic dexamethasone, which are presumably acting on the hippocampus. Also, lesions of the nucleus accumbens block the memory-enhancing effects of intra-amygdala infusions of glucocorticoid receptor agonist. Finally, the cortex is also a locus for memory consolidation, since projections from the basal lateral amygdala are essential in the modulation of memory by the entorhinal cortex (165, 170).
These results support the concept that CRH, by means of an interaction with glucocorticoids, interacts with the noradrenergic system to consolidate traumatic memories. Individuals with excessive stress-induced release of CRH, cortisol, and norepinephrine are likely to be prone to the development of indelible traumatic memories and their associated reexperiencing symptoms. Administration of CRH antagonists, glucocorticoid receptor antagonists, and β-adrenergic receptor antagonists may prevent these effects in vulnerable subjects.
Reconsolidation
Reconsolidation is a process in which old, reactivated memories undergo another round of consolidation (171–173). The process of reconsolidation is extremely relevant to both vulnerability and resiliency to the effects of extreme stress. It is the rule rather than the exception that memories are reactivated by cues associated with the original trauma. Repeated reactivation of these memories may serve to strengthen the memories and facilitate long-term consolidation (174, 175). Each time a traumatic memory is retrieved, it is integrated into an ongoing perceptual and emotional experience and becomes part of a new memory. Moreover, preclinical studies indicate that consolidated memories for auditory fear conditioning, which are stored in the amygdala (176), hippocampal-dependent contextual fear memory (171), and hippocampal-dependent memory associated with inhibitory avoidance (172) are sensitive to disruption upon reactivation by administration of a protein synthesis inhibitor directly into the amygdala and hippocampus, respectively. The reconsolidation process, which has enormous clinical implications, results in reactivated memory trace then returns to a state of lability and must undergo consolidation once more if it is to remain in long-term storage. Some controversies persist regarding the temporal persistence of systems reconsolidation. Debiec and colleagues (171) found that intrahippocampal infusions of anisomycin caused amnesia for a consolidated hippocampal-dependent memory if the memory was reactivated, even up to 45 days after training. Milekic and Alberini (172) however, found that the ability of intrahippocampal infusion of anisomycin to produce amnesia for an inhibitory avoidance task was evident only when the memory was recent (up to 7 days old). Further work is needed to resolve this very important question (173).
The reconsolidation process involves NMDA receptors and β-adrenergic receptors and requires cAMP response-element binding protein induction. The cAMP response- element binding protein requirement suggests that nuclear protein synthesis is necessary (177). NMDA receptor antagonists and β receptor antagonists impair reconsolidation (174, 178). The effect of the β receptor antagonist propranolol was greater after memory reactivation than when administered immediately after the initial training. These results suggest that reactivation of memory initiates a cascade of intracellular events that involve both NMDA receptor and β receptor activation in a fashion similar to postacquisition consolidation.
This remarkable lability of a memory trace, which permits a reorganization of an existing memory in a retrieval environment, provides a theoretical basis for both psychotherapeutic and pharmacotherapeutic intervention for traumatic stress exposure. Administration of β receptor and NMDA receptor antagonists shortly after the initial trauma exposure as well as after reactivation of memory associated with the event may reduce the strength of the original traumatic memory.
Extinction
When the conditioned stimuli are presented repeatedly in the absence of the unconditioned stimuli, a reduction in the conditioned fear response occurs. This process is called extinction. It forms the basis for exposure-based psychotherapies for the treatment of a variety of clinical conditions characterized by exaggerated fear responses. Individuals who show an ability to attenuate learned fear quickly through powerful and efficient extinction processes are likely to function more effectively under dangerous conditions. They may also be less susceptible to the effects of intermittent exposure to fear stimuli, which can reinstate fear-conditioned learning. Highly stress-resilient individuals under extreme stress generally experience fear but have the capacity to function well under states of high fear. In addition, individuals in positions that regularly cause them to confront danger need to be able to extinguish learned fears rapidly.
Extinction is characterized by many of the same neural mechanisms as in fear acquisition. Activation of amygdala NMDA receptors by glutamate is essential (179), and L-type voltage-gated calcium channels also contribute to extinction plasticity (180). Long-term extinction memory is altered by a number of different neurotransmitter systems, including GABA, norepinephrine, and dopamine, in a manner similar to fear acquisition (181, 182).
Destruction of the medial prefrontal cortex blocks recall of fear extinction (183, 184), indicating that the medial prefrontal cortex might store long-term extinction memory. Infralimbic neurons, which are part of the medial prefrontal cortex, fire only when rats are recalling extinction—greater firing correlates with reduced fear behaviors (185). It has been suggested that the consolidation of extinction involves potentiation of inputs into the medial prefrontal cortex by means of NMDA-dependent plasticity. The basal lateral amygdala sends direct excitatory inputs to the medial prefrontal cortex, and NMDA antagonists infused into the basal lateral amygdala block extinction. The ability of the medial prefrontal cortex to modulate fear behaviors is probably related to projections from the medial prefrontal cortex by means of GABA interneurons to the basal lateral amygdala (186).
Failure to achieve an adequate level of activation of the medial prefrontal cortex after extinction might lead to persistent fear responses (187). Individuals with the capacity to function well after experiencing states of high fear may have potent medial prefrontal cortex inhibition of amygdala responsiveness. In contrast, patients with PTSD exhibit depressed ventral medial prefrontal cortex activity, which correlates with increased autonomic arousal after exposure to traumatic reminders (unpublished work by Bremner et al.). Consistent with this hypothesis, we (188) showed that PTSD patients had increased left amygdala activation during fear acquisition and decreased activity of the medial prefrontal cortex/anterior cingulate during extinction. It has been proposed that potentiating NMDA receptors using the glycine agonist d-cycloserine may facilitate the extinction process when given in combination with behavioral therapy in patients with anxiety disorders (189).
The Neural Basis of Social Behavior
As noted, a number of scholars have worked to define the psychological characteristics that promote resiliency. These characteristics include being altruistic toward others and having the ability to attract and use support (139–141). Therefore, understanding the neural basis of altruism and other forms of adaptive social behavior may be relevant to a better conceptualization of the psychobiology of resilience.
Preclinical studies have used several rodent model systems to increase our knowledge of how the brain processes social information and regulates social behavior (190). These models include the oxytocin knockout mouse and the study of the neurobiology of social behaviors in prairie and montane voles. The oxytocin knockout mouse exhibits a specific deficit in social recognition in the context of intact general cognitive abilities and olfactory processing (191). Social recognition is fully restored by oxytocin infusion during the initial processing of social information. Studies measuring C-Fos induction indicate that the medial amygdala is involved in pathways that differentially process social and nonsocial information (192).
Prairie and montane voles are similar genetically but vary greatly in their social behaviors. The prairie vole is highly social, forms long-lasting social attachments, and is monogamous (193), whereas the montane vole avoids social contact except for the purpose of mating (194). Oxytocin and vasopressin appear to play crucial roles in the social behavior of prairie voles. They increase the amount of time a vole spends socially engaged and are involved in the formation of the pair bond. The levels of oxytocin and vasopressin are similar in prairie and montane voles. The differences in social behavior are explained by the regional expression of these peptides in the brain. Prairie voles have high levels of oxytocin receptors in the nucleus accumbens and the basal lateral amygdala relative to montane voles (195). Similarly, prairie voles have higher densities of the vasopressin-1A receptor on the ventral pallidum and the medial amygdala than montane voles. Infusion of vasopressin has different effects in the two voles; prairie voles increase social interaction, and montane voles increase nonsocial behaviors, such as autogrooming (196). The neural mechanisms responsible for the effects of oxytocin and vasopressin on social behavior are thought to involve some of the same circuitry (the nucleus accumbens and the ventral pallidum) involved in reward-related behavior. These brain regions are also components of the dopamine reward system (197). This suggests that, in prairie voles, activation of these brain regions during social interactions reinforces social behavior.
Recently, there have been several human studies that bear on the neural basis of social cooperation. Reciprocal altruism is a core behavioral principle of human social life and has been related to resilience. Rilling and colleagues (198) studied social cooperation with the iterated Prisoner’s Dilemma Game. They found that mutual cooperation was associated with consistent activation of brain areas linked to reward processing, including the nucleus accumbens, the caudate nucleus, and regions of the medial prefrontal cortex. They hypothesized that this pattern of neural activation by means of linkage to reward circuits sustains cooperative social relationships and inhibits the selfish impulse to accept but not to reciprocate an act of altruism. Highly resilient children, adolescents, and adults have exceptional abilities to form supportive social attachments. Individuals who demonstrate outstanding leadership ability and courageous acts in the context of great personal danger are frequently characterized by unique altruism. Clinical studies in such individuals designed to examine the neural circuits related to social cooperation are now indicated.
Future Research Directions
Examination of the neural circuits of reward, fear conditioning and extinction, and social behavior reveal that several brain structures are involved in more than one circuit (Figure 2). This is most striking for the amygdala, the nucleus accumbens, and the medial prefrontal cortex. The amygdala has been most prominently identified as a critical structure in studies of fear conditioning; however, it also has a major role in reward mechanisms. The nucleus accumbens is implicated in both reward and social behaviors, and the medial prefrontal cortex is a component of all three circuits.
These observations raise many intriguing questions. For example, does a particular level of amygdala function in fear conditioning relate in a predictable way to its function in the reward system? Does the finding of increased amygdala responsiveness to fear stimuli in PTSD and depression suggest that amygdala dysfunction will also be apparent in the study of reward in these disorders? To carry this a step further, will there be a clinical correlation between abnormalities in fear regulation and anhedonia? The redundancy in the circuits mediating reward and social behavior, especially involving the nucleus accumbens, suggests a functional interaction between these two circuits. When both systems are functioning well, positive social behaviors are reinforced. However, an inability to experience reward because of an impaired circuit may result in unrewarding social experiences, deficient social competence, and social withdrawal. The medial prefrontal cortex is believed to be a critical link between emotional regulation and higher-level decision making. Abnormalities in functioning of the medial prefrontal cortex could be manifested by a failure to assess accurately the range of outcomes associated with reward or punishment, indicative of a functional relationship between reward and fear circuits. Moreover, it has been suggested that this region regulates social interactions, including the capacity for empathy and altruism (199).
To date, most neuroimaging studies have investigated the functional status of these circuits in isolation and not in relation to each other. This article suggests that assessment of the functional relationships among these circuits, including the associated neurochemical modulation, may be important in providing a more comprehensive and precise understanding of the contribution of these circuits to resilience and vulnerability to stress.
There is emerging evidence indicating that genetic factors contribute to the vulnerability to stress-related psychopathology, such as in PTSD. An investigation of twin pairs from the Vietnam Twin Registry (200) reported that inherited factors accounted for up to 32% of the variance of PTSD symptoms beyond the contribution of trauma severity. The molecular neurobiological abnormalities that underlie these findings have not been elucidated. Two relatively small association studies (201, 202) that evaluated dopamine D2 receptor polymorphisms in PTSD yielded contradictory results. A preliminary study (203) found an association between the dopamine transporter polymorphism and PTSD. Volumetric magnetic resonance imaging investigations (204–206) demonstrated a smaller hippocampal volume in PTSD patients. A study of monozygotic twins discordant for trauma exposure (207) found evidence that smaller hippocampal volume may constitute a risk factor for the development of stress-related psychopathology. The recent identification of functional polymorphisms for the glucocorticoid receptor (208), for the α2C adrenergic receptor subtype (209), and for neuropeptide Y synthesis (210) provides opportunities to investigate the genetic basis of the neurochemical response pattern to stress.
Work is commencing to examine the genetic basis of the neural mechanisms of reward, fear conditioning, and social behavior. There have been several recent advances in understanding the genetic contribution and molecular machinery related to amygdala-dependent learned fear. A gene-encoding gastrin-releasing peptide has been identified in the lateral amygdala. The gastrin-releasing peptide receptor is expressed in GABA-ergic interneurons and mediates their inhibition of principal neurons. In knockout mice with the gastrin-releasing peptide receptor, this inhibition is reduced and long-term potentiation is enhanced. These mice have enhanced and prolonged fear memory for auditory and contextual cues, indicating that the gastrin-releasing peptide signaling pathway may serve as an inhibitory feedback constraint on learned fear (159). The work further supports a role of GABA in fear and anxiety states (211) and suggests the genetic basis of vulnerability to anxiety may relate to gastrin-releasing peptide, gastrin-releasing peptide receptor, and GABA (212). Other preclinical studies indicate that there may be a genetically determined mesocortical and mesoaccumbens dopamine response to stress that relates to learned helplessness (80). There may be genetic mechanisms affecting social affiliation behavior that involves the vasopressin-1A receptor that can be evaluated in clinical populations (213). Recently, it was demonstrated that healthy subjects with the serotonin transporter polymorphism that has been associated with reduced 5-HT expression and function and increased fear and anxiety behaviors exhibit increased amygdala neuronal activity in response to fear-inducing stimuli (214–216). These preclinical and clinical data suggest that multidisciplinary studies that use neurochemical, neuroimaging, and genetic approaches have the potential to clarify the complex relationships among genotype, phenotype, and psychobiological responses to stress.
Philosophers and behavioral scientists have been interested in stress, coping, and resilience since antiquity, such as when Socrates said to Laches, “Tell me, if you can, what is courage” (217). Epidemiological and phenomenological investigations reveal specific individual, familial, and community characteristics that promote resilience and have even informed social policy. We now have the opportunity to bring to bear the full power of advances in our understanding of the neurobiological basis of behavior, to break down the artificial boundaries of mind-brain and nature-nurture (218), and to create more comprehensive psychobiological models of what Ann Masten has termed the “ordinary magic” of resilience processes (219). Such models will facilitate badly needed discoveries that will enhance our ability to predict, prevent, and treat stress-related psychopathology.
 |
 |
Received Feb. 19, 2003; revision received June 3, 2003; accepted June 17, 2003. From the Mood and Anxiety Disorders Program, NIMH. Address reprint requests to Dr. Charney, Mood and Anxiety Disorders Program, NIMH, 15K North Dr., Rm. 101, Bethesda, MD 20892-2670; [email protected] (e-mail).

Figure 1. Neurochemical Response Patterns to Acute Stressa
aThis figure illustrates some of the key brain structures involved in the neurochemical response patterns following acute psychological stress. The functional interactions among the different neurotransmitters, neuropeptides, and hormones are emphasized. It is apparent the functional status of brain regions such as the amygdala (neuropeptide Y, galanin, corticotropin-releasing hormone [CRH], cortisol, and norepinephrine), hippocampus (cortisol and norepinephrine), locus coeruleus (neuropeptide Y, galanin, and CRH), and prefrontal cortex (dopamine, norepinephrine, galanin, and cortisol) will depend upon the balance among multiple inhibitory and excitatory neurochemical inputs. It is also noteworthy that functional effects may vary depending on the brain region. Cortisol increases CRH concentrations in the amygdala and decreases concentrations in the paraventricular nucleus of the hypothalamus. As described in the text, these neurochemical response patterns may relate to resilience and vulnerability to the effects of extreme psychological stress.

Figure 2. Neural Circuits Associated With Reward, Fear Conditioning, and Social Behaviora
aThe figure depicts a simplified summary of some of the brain structures and relevant neurochemistry mediating the neural mechanisms of reward (purple paths), fear conditioning and extinction (yellow paths), and social behaviors (blue paths). Only a subset of the many known interconnections among these various regions is shown, and relevant interneurons are not illustrated (see text), yet it can be seen there is considerable overlap in the brain structures associated with these neural mechanisms. This suggests that there may be clinically relevant functional interactions among the circuits. For example, a properly functioning reward circuit may be necessary for the reinforcement of positive social behaviors. An overly responsive fear circuit or impaired extinction process may negatively influence functioning of the reward system. The assessment of these neural mechanisms must be considered in the context of their neurochemical regulation. Alterations in one neurotransmitter, neuropeptide, or hormone system will affect more than one circuit. Several receptors that are related to putative antianxiety and antidepressant drug targets are illustrated. The functional status of these circuits has important influences on stress-related psychopathology and the discovery of novel therapeutics (see text).
1. Sterling P, Eyer J: Allostasis: a new paradigm to explain arousal pathology, in Handbook of Life Stress, Cognition, and Health. Edited by Fisher S, Reason J. New York, John Wiley & Sons, 1988, pp 629–649Google Scholar
2. McEwen BS: Sex, stress, and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging 2002; 23:921–939Crossref, Medline, Google Scholar
3. McEwen BS, Stellar E: Stress and the individual: mechanisms leading to disease. Arch Intern Med 1993; 153:2093–2101Crossref, Medline, Google Scholar
4. Gold PW, Drevets WC, Charney DS: New insights into the role of cortisol and the glucocorticoid receptor in severe depression. Biol Psychiatry 2002; 52:381–385Crossref, Medline, Google Scholar
5. Makino S, Gold PW, Schulkin J: Effects of corticosterone on CRH mRNA and content in the bed nucleus of the stria terminalis; comparison with the effects in the central nucleus of the amygdala and the paraventricular nucleus of the hypothalamus. Brain Res 1994; 675:141–149Crossref, Google Scholar
6. Makino S, Gold PW, Schulkin J: Corticosterone effects on corticotropin-releasing hormone mRNA in the central nucleus of the amygdala and the parvocellular region of the paraventricular nucleus of the hypothalamus. Brain Res 1994; 640:105–112Crossref, Medline, Google Scholar
7. Shepard JD, Barron KW, Myers DA: Corticosterone delivery to the amygdala increases corticotropin-releasing factor mRNA in the central amygdaloid nucleus and anxiety-like behavior. Brain Res 2000; 861:288–295Crossref, Medline, Google Scholar
8. Lee Y, Schulkin J, Davis M: Effect of corticosterone on the enhancement of the acoustic startle reflex by corticotropin releasing factor (CRF). Brain Res 1994; 666:93–98Crossref, Medline, Google Scholar
9. Roozendaal B: Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology 2000; 25:213–238Crossref, Medline, Google Scholar
10. Diamond DM, Fleshner M, Ingersoll N, Rose GM: Psychological stress impairs spatial working memory: relevance to electrophysiological studies of hippocampal function. Behav Neurosci 1996; 110:661–672Crossref, Medline, Google Scholar
11. Karlamangla AS, Singer BH, McEwen BS, Rowe JW, Seeman TE: Allostatic load as a predictor of functional decline: MacArthur studies of successful aging. J Clin Epidemiol 2002; 55:696–710Crossref, Medline, Google Scholar
12. Rosenfeld RS, Hellman L, Roffwarg H, Weitzman ED, Fukushima DK, Gallagher TF: Dehydroisoandrosterone is secreted episodically and synchronously with cortisol by normal man. J Clin Endocrinol Metab 1971; 33:87–92Crossref, Medline, Google Scholar
13. Browne ES, Wright BE, Porter JR, Svec F: Dehydroepiandrosterone: antiglucocorticoid action in mice. Am J Med Sci 1992; 303:366–371Crossref, Medline, Google Scholar
14. Rose KA, Stapleton G, Dott K, Kieny MP, Best R, Schwarz M, Russell DW, Bjorkheim I, Seckl J, Lathe R: Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7alpha-hydroxy dehydroepiandrosterone and 7alpha-hydroxy pregnenolone. Proc Natl Acad Sci USA 1997; 94:4925–4930Crossref, Medline, Google Scholar
15. Morfin R, Starka L: Neurosteroid 7-hydroxylation products in the brain. Int Rev Neurobiol 2001; 46:79–95Crossref, Medline, Google Scholar
16. Kimonides VG, Khatibi NH, Svendsen CN, Sofroniew MV, Herbert J: Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc Natl Acad Sci USA 1998; 95:1852–1857Crossref, Medline, Google Scholar
17. Bastianetto S, Ramassamy C, Poirier J, Quirion R: Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Brain Res Mol Brain Res 1999; 66:35–41Crossref, Medline, Google Scholar
18. Kaminska M, Harris J, Gilsbers K, Dubrovsky B: Dehydroepiandrosterone sulfate (DHEAS) counteracts decremental effects of corticosterone on dentate gyrus LTP: implications for depression. Brain Res Bull 2000; 52:229–234Crossref, Medline, Google Scholar
19. Goodyer IM, Herbert J, Altham PME: Adrenal steroid secretion and major depression in 8- to 16-year-olds, III: influence of cortisol/DHEA ratio at presentation on subsequent rates of disappointing life events and persistent major depression. Psychol Med 1998; 28:265–273Crossref, Medline, Google Scholar
20. Goodyer IM, Park RJ, Netherton CM, Herberg J: Possible role of cortisol and dehydroepiandrosterone in human development and psychopathology. Br J Psychiatry 2001; 179:243–249Crossref, Medline, Google Scholar
21. Young AH, Gallagher P, Porter RJ: Elevation of the cortisol-dehydroepiandrosterone ratio in drug-free depressed patients. Am J Psychiatry 2002; 159:1237–1239Link, Google Scholar
22. Wolkowitz OM, Reus VI, Keebler A, Nelson N, Friedland M, Brizendine L, Roberts E: Double-blind treatment of major depression with dehydroepiandrosterone. Am J Psychiatry 1999; 156:646–649Abstract, Google Scholar
23. Majewska MD: Neurosteroids: endogenous bimodal modulators of the GABAA receptor: mechanism of action and physiological significance. Prog Neurobiol 1992; 38:379–395Crossref, Medline, Google Scholar
24. Bergeron R, de Montigny C, Debonnel G: Potentiation of neuronal NMDA response induced by dehydroepiandrosterone and its suppression by progesterone: effects mediated by sigma receptors. J Neurosci 1996; 16:1193–1202Crossref, Medline, Google Scholar
25. Grammatopoulos DK, Chrousos GP: Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol Metab 2002; 13:436–444Crossref, Medline, Google Scholar
26. Steckler T, Holsboer F: Corticotropin-releasing hormone receptor subtypes and emotion. Biol Psychiatry 1999; 46:1480–1508Crossref, Medline, Google Scholar
27. Strome EM, Trevor GHW, Higley JD, Liriaux DL, Suomi SJ, Doudet DJ: Intracerebroventricular corticotropin-releasing factor increased limbic glucose metabolism and has social context dependent behavioral effects in nonhuman primates. Proc Natl Acad Sci USA 2002; 99:15749–15754Crossref, Medline, Google Scholar
28. Nemeroff CB: Recent advances in the neurobiology of depression. Psychopharmacol Bull 2002; 36:6–23Medline, Google Scholar
29. Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS: Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997; 154:624–629Link, Google Scholar
30. Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD Jr: Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999; 156:585–588; correction, 156:986Abstract, Google Scholar
31. Sanchez MM, Young LJ, Plotsky PM, Insel TR: Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J Comp Neurol 1999; 408:365–377Crossref, Medline, Google Scholar
32. Bale TL, Picetti R, Contarino A, Koob GF, Vale WW, Kuo-Fen L: Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior. J Neurosci 2002; 22:193–199Crossref, Medline, Google Scholar
33. Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF: Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behavior and are hypersensitive to stress. Nat Genet 2000; 24:410–414Crossref, Medline, Google Scholar
34. Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR, Hatton DC, Phillips TJ, Finn DA, Low MJ, Rittenberg MB, Stenzel P, Stenzel-Poore MP: Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet 2000; 24:403–409Crossref, Medline, Google Scholar
35. Charney DS, Bremner JD: The neurobiology of anxiety disorders, in Neurobiology of Mental Illness. Edited by Charney DS, Nestler EJ, Bunney BS. New York, Oxford University Press, 1999, pp 494–517Google Scholar
36. Charney DS, Woods SW, Goodman WK, Heninger GR: Neurobiological mechanisms of panic anxiety: biochemical and behavioral correlates of yohimbine-induced panic attacks. Am J Psychiatry 1987; 144:1030–1036Link, Google Scholar
37. Charney DS, Woods SW, Krystal JH, Nagy LM, Heninger GR: Noradrenergic neuronal dysregulation in panic disorder: the effects of intravenous yohimbine and clonidine in panic disorder patients. Acta Psychiatr Scand 1992; 86:273–282Crossref, Medline, Google Scholar
38. Geracioti TD Jr, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, Schmidt D, Rounds-Kugler B, Yehuda R, Keck PE Jr, Kasckow JW: CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry 2001; 158:1227–1230Link, Google Scholar
39. Southwick SM, Krystal JH, Bremner JD, Morgan CA, Nicolaou AL, Nagy LM, Johnson DR, Heninger GR, Charney DS: Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry 1997; 4:749–758Crossref, Google Scholar
40. Wong ML, Kling MA, Munson PJ, Listwak S, Licinio J, Prolo P, Karp B, McCutcheon IE, Geracioti TD Jr, DeBellis MD, Rice KC, Goldstein DS, Veldhuis JD, Chrousos GP, Oldfield EH, McCann SM, Gold PW: Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci USA 2000; 97:325–330Crossref, Medline, Google Scholar
41. Makino S, Baker RA, Smith MA, Gold PW: Differential regulation of neuropeptide Y mRNA expression in the accurate nucleus and locus coeruleus by stress and antidepressants. J Neuroendocrinol 2000; 12:387–395Crossref, Medline, Google Scholar
42. Baker RA, Herkenham M: Arcuate nucleus neurons that project to the hypothalamic paraventricular nucleus: neuropeptidergic identity and consequences of adrenalectomy on mRNA levels in the rat. J Comp Neurol 1995; 358:518–530Crossref, Medline, Google Scholar
43. Risold PY, Swanson LW: Chemoarchitecture of the rat lateral septal nucleus. Brain Res Brain Res Rev 1997; 24:91–113Crossref, Medline, Google Scholar
44. Pieribone VA, Brodin L, Friberg K, Dahlstrand J, Soderberg C, Larhammar D, Hokfelt T: Differential expression of mRNAs for neuropeptide Y-related peptides in rat nervous tissues: possible evolutionary conservation. J Neurosci 1992; 12:3361–3371Crossref, Medline, Google Scholar
45. Allen YS, Adrian TE, Allen JM, Tatemoto K, Crow TJ, Bloom SR, Polak JM: Neuropeptide Y distribution in the rat brain. Science 1983; 221:877–879Crossref, Medline, Google Scholar
46. Heilig M, McLeod S, Brot M, Heinrichs SC, Menzaghi F, Koob GF, Britton KT: Anxiolytic-like action of neuropeptide Y: mediation by Y1 receptors in amygdala, and dissociation from food intake effects. Neuropsychopharmacology 1993; 8:357–363Crossref, Medline, Google Scholar
47. Heilig M: Antisense inhibition of neuropeptide Y (NPY)-Y1 receptor expression blocks the anxiolytic-like action of NPY in amygdala and paradoxically increases feeding. Regul Pept 1995; 59:201–205Crossref, Medline, Google Scholar
48. Sajdyk TJ, Vandergriff MG, Gehlert DR: Amygdalar neuropeptide Y Y1 receptors mediate the anxiolytic-like actions of neuropeptide Y in the social interaction test. Eur J Pharmacol 1999; 368:143–147Crossref, Medline, Google Scholar
49. Thorsell A, Carlsson K, Ekman R, Heilig M: Behavioral and endocrine adaptation, and up-regulation of NPY expression in rat amygdala following repeated restraint stress. Neuroreport 1999; 10:3003–3007Crossref, Medline, Google Scholar
50. Flood JF, Baker ML, Hernandez EN, Morley JE: Modulation of memory processing by neuropeptide Y varies with brain injection site. Brain Res 1989; 503:73–82Crossref, Medline, Google Scholar
51. Illes P, Finta EP, Nieber K: Neuropeptide Y potentiates via Y2-receptors the inhibitory effect of noradrenaline in rat locus coeruleus neurons. Naunyn Schmiedebergs Arch Pharmacol 1993; 348:546–548Crossref, Medline, Google Scholar
52. Thorsell A, Michalkiewicz M, Dumont Y, Quirion R, Caberlotto L, Rimondini R, Mathe AA, Helig M: Behavioral insensitivity to restraint stress, absent fear suppression of behavior, and impaired spatial learning in transgenic rats with hippocampal neuropeptide Y overexpression. Proc Natl Acad Sci USA 2000; 97:12852–12857Crossref, Medline, Google Scholar
53. Britton KT, Akwa Y, Spina MG, Koob GF: Neuropeptide Y blocks anxiogenic-like behavioral action of corticotropin-releasing factor in an operant conflict test and elevated plus maze. Peptides 2000; 21:37–44Crossref, Medline, Google Scholar
54. Heilig M, Koob GF, Ekman R, Britton KT: Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci 1994; 17:80–85Crossref, Medline, Google Scholar
55. Kask A, Rago L, Harro J: Alpha-helical CRF(9–41) prevents anxiogenic-like effect on NPY Y1 receptor antagonist BIBP3226 in rats. Neuroreport 1997; 8:3645–3647Crossref, Medline, Google Scholar
56. Kask A, Harro J, von Horsten S, Redrobe JP, Dumont Y, Quiron R: The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci Behav Rev 2002; 26:259–283Crossref, Medline, Google Scholar
57. Smagin GN, Harris RB, Ryan DH: Corticotropin-releasing factor receptor antagonist infused into the locus coeruleus attenuates immobilization stress-induced defensive withdrawal in rats. Neurosci Lett 1996; 220:167–170Crossref, Medline, Google Scholar
58. Kask A, Rago L, Harro J: Naxiolytic-like effect of neuropeptide Y (NPY) and NPY 13–36 microinjected into vicinity of locus coeruleus in rats. Brain Res 1998; 788:345–348Crossref, Medline, Google Scholar
59. Sheriff S, Dautzenberg FM, Mulchahey JJ, Pisarska M, Hauger RL, Chance WT, Balasubramaniam A, Kasckow JW: Interaction of neuropeptide Y and corticotropin-releasing factor signaling pathways in AR-5 amygdalar cells. Peptides 2001; 22:2083–2089Crossref, Medline, Google Scholar
60. Sajdyk TJ, Schober DA, Gehlert DR, Shekhar A: Role of corticotropin-releasing factor and urocortin within the basolateral amygdala of rats in anxiety and panic responses. Behav Brain Res 1999; 100:207–215Crossref, Medline, Google Scholar
61. Kask A, Rago L, Harro J: NPY Y1 receptors in the dorsal periaqueductal gray matter regulate anxiety in the social interaction test. Neuroreport 1998; 9:2713–2716Crossref, Medline, Google Scholar
62. Martins AP, Maras RA, Guimaraes FS: Anxiolytic effect of a CRH receptor antagonist in the dorsal periaqueductal gray. Depress Anxiety 2001; 12:99–101Crossref, Google Scholar
63. Morgan CA, Wang S Mason J, Southwick SM, Fox P, Hazlett G, Charney DS, Greenfield G: Hormone profiles in humans experiencing military survival training. Biol Psychiatry 2000; 47:891–901Crossref, Medline, Google Scholar
64. Rasmusson AM, Hauger RI, Morgan CA, Bremner JD, Charney DS, Southwick SM: Low baseline and yohimbine-stimulated plasma neuropeptide Y (NPY) levels in combat-related PTSD. Biol Psychiatry 2000; 47:526–539Crossref, Medline, Google Scholar
65. Mathe HH: Early life stress changes concentrations of neuropeptide Y and corticotropin-releasing hormone in adult rat brain: lithium treatment modifies these changes. Neuropsychopharmacology 2002; 27:756–764Crossref, Medline, Google Scholar
66. Holmes A, Yang RJ, Crawley JN: Evaluation of an anxiety-related phenotype in galanin overexpressing transgenic mice. J Mol Neurosci 2002; 18:151–165Crossref, Medline, Google Scholar
67. Holmes PV, Crawley JN: Coexisting neurotransmitters in central noradrenergic neurons, in Psychopharmacology: The Fourth Generation of Progress. Edited by Bloom FE, Kupfer DJ. New York, Raven Press, 1995, pp 347–353Google Scholar
68. Gentleman SM, Falkai P, Bogerts B, Herrero MT, Polak JM, Roberts GW: Distribution of galanin-like immunoreactivity in the human brain. Brain Res 1989; 505:311–315Crossref, Medline, Google Scholar
69. Perez SE, Wynic D, Steiner RA, Mufson EJ: Distribution of galaninergic immunoreactivity in the brain of the mouse. J Comp Neurol 2001; 434:158–185Crossref, Medline, Google Scholar
70. Sevcik J, Finta EP, Illes P: Galanin receptors inhibit the spontaneous firing of locus coeruleus neurons and interact with mu-opioid receptors. Eur J Pharmacol 1993; 230:223–230Crossref, Medline, Google Scholar
71. Xu ZQ, Tong YG, Hokfelt T: Galanin enhances noradrenaline-induced outward current on locus coeruleus noradrenergic neurons. Neuroreport 2001; 12:1179–1182Google Scholar
72. Bing O, Moller C, Engel JA, Soderpal B, Heilig M: Anxiolytic-like action of centrally administered galanin. Neurosci Lett 1993; 164:17–20Crossref, Medline, Google Scholar
73. Moller C, Sommer W, Thorsell A, Heilig M: Anxiogenic-like action of galanin after intra-amygdala administration in the rat. Neuropsychopharmacology 1999; 21:507–512Crossref, Medline, Google Scholar
74. Khoshbouei H, Cecchi M, Dove S, Javors M, Morilak DA: Behavioral reactivity to stress: amplification of stress-induced noradrenergic activation elicits a galanin-mediated anxiolytic effect in central amygdala. Pharmacol Biochem Behav 2002; 71:407–417Crossref, Medline, Google Scholar
75. Kinney JW, Starosta G, Holmes A, Wrenn CC, Yang RJ, Harris AP, Long KC, Crawley JN: Deficits in trace cued fear conditioning in galanin-treated rats and galanin-overexpressing transgenic mice. Learn Mem 2002; 9:178–190Crossref, Medline, Google Scholar
76. Gustafson EL, Smith KE, Durkin MM, Gerald C, Branchek TA: Distribution of a rat galanin receptor mRNA in rat brain. Neuroreport 1996; 7:953–957Crossref, Medline, Google Scholar
77. Holmes A, Kinney JW, Wrenn CC, Li Q, Yang RJ, Ma L, Vishwanath J, Saavedra MC, Innerfield CE, Jacoby AS, Shine J, Iismaa TP, Crawley JN: Galanin GAL-R1 receptor null mutant mice display increased anxiety-like behavior specific to the elevated plus-maze. Neuropsychopharmacology 2003; 28:1931–1044Google Scholar
78. Hokfelt T, Millhorn D, Seroogy K, Seroogy K, Tsuruo Y, Ceccatelli S, Lindh B, Meister B, Melander T, Schalling M, Bartfai T, et al: Coexistence of peptides with classical transmitters. Experientia 1987; 43:768–780Crossref, Medline, Google Scholar
79. Consolo S, Baldi G, Russi G, Civenni G, Bartfai T, Vezzani A: Impulse flow dependency of galanin release in vivo in the rat ventral hippocampus. Proc Natl Acad Sci USA 1994; 91:8047–8051Crossref, Medline, Google Scholar
80. Ventura R, Cabib S, Puglisi-Allegra S: Genetic susceptibility of mesocortical dopamine to stress determines liability to inhibition of mesoaccumbens dopamine and to behavioral despair in a mouse model of depression. Neuroscience 2002; 115:99–1007Crossref, Google Scholar
81. Cabib S, Puglisi-Allegra S: Different effects of repeated stressful experiences on mesocortical and mesolimbic dopamine metabolism. Neuroscience 1996; 73:375–380Crossref, Medline, Google Scholar
82. Cabib S, Ventgura R, Puglisi-Allegra S: Opposite imbalances between mesocortical and mesoaccumbens dopamine responses to stress by the same genotype depending on living conditions. Behav Brain Res 2002; 129:179–185Crossref, Medline, Google Scholar
83. Goldstein LE, Rasmusson AM, Bunney BS, Roth RH: Role of the amygdala in the coordination of behavioral, neuroendocrine, and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci 1996; 16:4787–4798Crossref, Medline, Google Scholar
84. Morrow BA, Elsworth JD, Rasmusson AM, Roth RH: The role of mesoprefrontal dopamine neurons in the acquisition and expression of conditioned fear in the rat. Neuroscience 1999; 92:553–564Crossref, Medline, Google Scholar
85. Lemieux AM, Coe CL: Abuse-related PTSD: evidence for chronic neuroendocrine activation in women. Psychosom Med 1995; 57:105–115Crossref, Medline, Google Scholar
86. Hamner MB, Diamond BI: Elevated plasma dopamine in posttraumatic stress disorder: a preliminary report. Biol Psychiatry 1993; 33:304–306Crossref, Medline, Google Scholar
87. Lambert G, Johansson M, Agren H, Friberg P: Reduced brain norepinephrine and dopamine release in treatment refractory depressive illness. Arch Gen Psychiatry 2000; 57:787–793Crossref, Medline, Google Scholar
88. Kent JM, Mathew SJ, Gorman JM: Molecular targets in the treatment of anxiety. Biol Psychiatry 2002; 52:1008–1030Crossref, Medline, Google Scholar
89. Charney DS, Drevets WD: Neurobiological basis of anxiety disorders, in Neuropsychopharmacology: The Fifth Generation of Progress. Edited by Davis KL, Charney D, Coyle JT, Nemeroff C. Philadelphia, Lippincott Williams & Wilkins, 2002Google Scholar
90. Pazos A, Probst A, Palacios J: Serotonin receptors in the human brain, III: autoradiographic mapping of serotonin-1 receptors. Neuroscience 1987; 21:97–122Crossref, Medline, Google Scholar
91. Hamon M, Gozlan H, el Mestikawy S, Emerit MB, Bolanos F, Schechter L: The central 5-HT1A receptors: pharmacological, biochemical, functional, and regulatory properties. Ann NY Acad Sci 1990; 600:114–129Crossref, Medline, Google Scholar
92. Heisler LK, Chu H-M, Brennan TJ, Danao JA, Bajwa P, Parsons LH, Tecott LH: Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc Natl Acad Sci USA 1998; 95:15049–15054Crossref, Medline, Google Scholar
93. Parks C, Robinson P, Sibille E, Shenk T, Toth M: Increased anxiety of mice lacking the serotonin 1A receptor. Proc Natl Acad Sci USA 1998; 95:10734–10739Crossref, Medline, Google Scholar
94. Gross C, Zhuang X, Stark K, Ramboz S, Oosting R, Kirby L, Santarelli L, Beck S, Hen R: Serotonin 1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature 2002; 416:396–400Crossref, Medline, Google Scholar
95. Lopez JF, Chalmers DT, Little KY, Watson SJ: AE Bennett Research Award: regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry 1998; 43:547–573Crossref, Medline, Google Scholar
96. Sibille E, Pavlides C, Benke D, Toth M: Genetic inactivation of the serotonin(1A) receptor in mice results in downregulation of major GABA(A) receptor alpha subunits, reduction of GABA(A) receptor binding, and benzodiazepine-resistant anxiety. J Neurosci 2000; 20:2758–2765Crossref, Medline, Google Scholar
97. Pattij T, Groenink L, Oosting RS, van der Gugten J, Maes RAA, Olivier B: GABA(A)-benzodiazepine receptor complex sensitivity in 5-HT(1A) receptor knockout mice on a 129/Sv background. Eur J Pharmacol 2002; 447:67–74Crossref, Medline, Google Scholar
98. Drevets WC, Frank JC, Kupfer DJ, Holt D, Greer PJ, Huang Y, Gautier C, Mathis C: PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry 1999; 46:1375–1387Crossref, Medline, Google Scholar
99. Neumeister A, Bain E, Nugent AC, Carson RE, Bonne O, Luckenbaugh DA, Eckelman W, Herscovitch P, Charney DS, Drevets W: Reduced serotonin type 1A receptor binding in panic disorder. J Neurosci (in press)Google Scholar
100. Weizman R, Weizman A, Kook KA, Vocci F, Deutsch S, Paul SM: Repeated swim stress alters brain benzodiazepine receptors measured in vivo. J Pharmacol Exp Ther 1989; 249:701–707Medline, Google Scholar
101. Nutt DJ, Malizia AL: New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. Br J Psychiatry 2001; 179:390–396Crossref, Medline, Google Scholar
102. Malizia AL, Cunningham VJ, Bell CJ, Liddle PF, Jones T, Nutt DJ: Decreased brain GABA(A)-benzodiazepine receptor binding in panic disorder: preliminary results from a quantitative PET study. Arch Gen Psychiatry 1998; 55:715–720Crossref, Medline, Google Scholar
103. Bremner JD, Innis RB, White T, Fujita M, Silbersweig D, Goddard AW, Staib L, Stern E, Cappiello A, Woods S, Baldwin R, Charney DS: SPECT [I-123]iomazenil measurement of the benzodiazepine receptor in panic disorder. Biol Psychiatry 2000; 47:96–106Crossref, Medline, Google Scholar
104. Bremner JD, Innis RB, Southwick SM, Staib L, Zoghbi S, Charney DS: Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am J Psychiatry 2000; 157:1120–1126Link, Google Scholar
105. Zitzmann M, Nieschlag E: Testosterone levels in healthy men and the relation to behavioural and physical characteristics: facts and constructs. Eur J Endocrinol 2001; 144:183–187Crossref, Medline, Google Scholar
106. Flugge G, Kramer M, Fuchs E: Chronic subordination stress in male tree shrews: replacement of testosterone affects behavior and central alpha2-adrenoceptors. Physiol Behav 2001; 73:293–300Crossref, Medline, Google Scholar
107. Schaal B, Tremblay RE, Soussignan R, Susman EJ: Male testosterone linked to high social dominance but low physical aggression in early adolescence. J Am Acad Child Adolesc Psychiatry 1996; 35:1322–1330Crossref, Medline, Google Scholar
108. Suay F, Salvador A, Gonzalez-Bono E, Sanchis C, Martinez M, Martinez-Sanchis S, Simon VM, Montoro JB: Effects of competition and its outcome on serum testosterone, cortisol and prolactin. Psychoneuroendocrinology 1999; 24:551–566Crossref, Medline, Google Scholar
109. Brooks JH, Reddon JR: Serum testosterone in violent and nonviolent young offenders. J Clin Psychol 1996; 52:475–485Crossref, Medline, Google Scholar
110. Banks T, Dabbs JM Jr: Salivary testosterone and cortisol in a delinquent and violent urban subculture. J Soc Psychol 1996; 136:49–56Crossref, Medline, Google Scholar
111. Aromaki AS, Lindman RE, Eriksson CJP: Testosterone aggressiveness and antisocial personality. Aggressive Behavior 1999; 25:113–123Crossref, Google Scholar
112. Lee S, Miselis R, Rivier C: Anatomical and functional evidence for a neural hypothalamic-testicular pathway that is independent of the pituitary. Endocrinology 2002; 143:4447–4454Crossref, Medline, Google Scholar
113. Mulchahey JJ, Ekhator NN, Zhang H, Kasckow JW, Baker DG, Geracioti TD Jr: Cerebrospinal fluid and plasma testosterone levels in post-traumatic stress disorder and tobacco dependence. Psychoneuroendocrinology 2001; 26:273–285Crossref, Medline, Google Scholar
114. Bauer M, Priebe S, Graef KJ, Keurten I: Psychosocial and endocrine abnormalities in refugees from East Germany, II: serum levels of cortisol, prolactin, luteinizing hormone, follicle stimulating hormone and testosterone. Psychiatry Res 1994; 51:75–85Crossref, Medline, Google Scholar
115. Pope HG Jr, Cohane GH, Kanayama G, Siegel AJ, Hudson JI: Testosterone gel supplementation for men with refractory depression: a randomized, placebo-controlled trial. Am J Psychiatry 2003; 160:105–111Link, Google Scholar
116. Wang C, Alexander G, Berman N, Salehian B, Davidson T, McDonald V, Steiner B, Hull L, Callegari C, Swerdloff RS: Testosterone replacement therapy improves mood in hypogonadal men—a clinical research center study. J Clin Endocrinol Metab 1996; 81:3578–3583Medline, Google Scholar
117. Stroud LR, Salovey P, Epel ES: Sex differences in stress responses: social rejection versus achievement stress. Biol Psychiatry 2002; 52:318–327Crossref, Medline, Google Scholar
118. Young EA, Altemus M, Parkison V, Shastry S: Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology 2001; 25:881–891Crossref, Medline, Google Scholar
119. Carey MP, Deterd CH, deKoning J, Helmerhorst F, de Kloet ER: The influence of ovarian steroids on hypothalamic-pituitary-adrenal regulation in the female rat. J Endocrinol 1995; 144:311–321Crossref, Medline, Google Scholar
120. Young EA: Sex differences in response to exogenous corticosterone. Mol Psychiatry 1996; 1:313–319Medline, Google Scholar
121. Komesaroff PA, Esler MD, Sudhir K: Estrogen supplementation attenuates glucocorticoid and catecholamine responses to mental stress in perimenopausal women. J Clin Endocrinol Metab 1999; 84:606–610Medline, Google Scholar
122. Cucinelli F, Soranna L, Barini A, Perri C, Leoni F, Mancuso S, Lanzone A: Estrogen treatment and body fat distribution are involved in corticotropin and cortisol response to corticotropin-releasing hormone in postmenopausal women. Metabolism 2002; 51:137–143Crossref, Medline, Google Scholar
123. Biller BM, Federoff JH, Loenig JL, Klibanski A: Abnormal cortisol secretion and responses to corticotropin-releasing hormone in women with hypothalamic amenorrhea. J Clin Endocrinol Metab 1990; 70:311–317Crossref, Medline, Google Scholar
124. Patchev VK, Hayashi S, Orikasa C, Almeida OF: Implications of estrogen-dependent brain organization for gender differences in hypothalamo-pituitary-adrenal regulation. FASEB J 1995; 9:419–423Crossref, Medline, Google Scholar
125. Hamlet MA, Rorie DK, Tyce GM: Effects of estradiol on release and disposition of norepinephrine from nerve endings. Am J Physiol 1980; 239:H450-H456Google Scholar
126. Colucci WS, Gimbrone MA, McLaughlin MK, Halpern W, Alexander RW: Increased vascular catecholamine sensitivity and alpha-adrenergic receptor affinity in female and estrogen-treated male rats. Circ Res 1982; 50:805–811Crossref, Medline, Google Scholar
127. Klangkalya B, Chan A: The effects of ovarian hormones on β-adrenergic and muscarinic receptors in rat heart. Life Sci 1988; 42:2307–2314Crossref, Medline, Google Scholar
128. Maggi A, Perez J: Estrogen-induced up-regulation of gamma-amino butyric receptors in the CNS of rodents. J Neurochem 1986; 47:1793–1797Crossref, Medline, Google Scholar
129. McEwen B: Estrogen actions throughout the brain. Recent Prog Horm Res 2002; 57:357–384Crossref, Medline, Google Scholar
130. Bethea CL, Mirkes SJ, Shively CA, Adams MR: Steroid regulation of tryptophan hydroxylase protein in the dorsal raphe of macaques. Biol Psychiatry 2000; 47:562–576Crossref, Medline, Google Scholar
131. Pecins-Thompson M, Brown NA, Bethea CL: Regulation of serotonin re-uptake transporter mRNA expression by ovarian steroids in rhesus macaques. Mol Brain Res 1998; 53:120–129Crossref, Medline, Google Scholar
132. Moses EL, Drevets WC, Smith G, Mathis CA, Kalro BN, Butters MA, Leondires MP, Greer PJ, Lopresti B, Loucks TL, Berga SL: Effects of estradiol and progesterone administration on human serotonin 2A receptor binding: a PET study. Biol Psychiatry 2000; 48:854–860Crossref, Medline, Google Scholar
133. Lu NZ, Bethea CL: Ovarian steroid regulation of 5-HT1A receptor binding and G protein activation in female monkeys. Neuropsychopharmacology 2002; 27:12–24Crossref, Medline, Google Scholar
134. Raap DK, DonCarlos L, Garcia F, Muma NA, Wolf WA, Battaglia G, Van de Kar LD: Estrogen desensitizes 5-HT(1A) receptors and reduces levels of G(z) G(i1) and G(i3) proteins in the hypothalamus. Neuropharmacology 2000; 39:1823–1832Crossref, Medline, Google Scholar
135. Mize AL, Poisner AM, Alper RH: Estrogens act in rat hippocampus and frontal cortex to produce rapid, receptor-mediated decreases in serotonin 5-HT1A receptor function. Neuroendocrinology 2001; 73:166–174Crossref, Medline, Google Scholar
136. Yonkers KA, Ellison JM: Anxiety disorders in women and their pharmacological treatment, in Psychopharmacology and Women. Edited by Jensvold MF, Halbreich U, Hamilton JA. Washington DC, American Psychiatric Press, 1996, pp 261–285Google Scholar
137. Seeman TE, Singer BH, Rowe JW, Horwitz RI, McEwen: Price of adaptation—allostatic load and its health consequences: MacArthur studies of successful aging. Arch Intern Med 1997; 157:2259–2268Crossref, Medline, Google Scholar
138. Seeman TE, McEwen BS, Rowe JW, Singer BH: Allostatic load as a marker of cumulative biological risk: MacArthur studies of successful aging. Proc Natl Acad Sci USA 2001; 98:4470–4475Crossref, Google Scholar
139. Richardson GE: The metatheory of resilience and resiliency. J Clin Psychol 2002; 58:307–321Crossref, Medline, Google Scholar
140. Masten AS, Coatsworth JD: The development of competence in favorable and unfavorable environments. Am Psychol 1998; 53:205–220Crossref, Medline, Google Scholar
141. Bell CC: Cultivating resiliency in youth. J Adolesc Health 2001; 29:375–381Crossref, Medline, Google Scholar
142. Grinker R, Spiegel J: Men Under Stress. Philadelphia, Blakiston, 1945Google Scholar
143. Rachman SJ: Fear and Courage, 2nd ed. New York, Freeman and Co, 1992Google Scholar
144. Ruff G, Korchin S: Psychological responses of the Mercury astronauts to stress, in The Threat of Impending Disaster. Edited by Grosser G, Wechsler H, Greenblatt M. Cambridge, Mass, MIT Press, 1964, pp 46–57Google Scholar
145. Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM: Neurobiology of depression. Neuron 2002; 34:13–25Crossref, Medline, Google Scholar
146. Wise RA: Brain reward circuitry: insights from unsensed incentives. Neuron 2002; 36:229–240Crossref, Medline, Google Scholar
147. Schultz W: Getting formal with dopamine and reward. Neuron 2002; 36:241–263Crossref, Medline, Google Scholar
148. Martin-Soelch C, Leenders KL, Chevalley AF, Missimer J, Kunig G, Magyar S, Mino A, Schultz W: Reward mechanisms in the brain and their role in dependence: evidence from neurophysiological and neuroimaging studies. Brain Res Rev 2001; 36:139–149Crossref, Medline, Google Scholar
149. Kotecha SA, Oak JN, Jackson MF, Perez Y, Orser BA, Van Tol HH, McDonald JF: A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron 2002; 35:1111–1122Crossref, Medline, Google Scholar
150. Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA Jr: Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated CREB expression in the nucleus accumbens. J Neurosci 2001; 21:7397–7403Crossref, Medline, Google Scholar
151. Hall J, Thomas KL, Everitt BJ: Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur J Neurosci 2001; 13:1453–1458Crossref, Medline, Google Scholar
152. Josselyn SA, Shi CJ, Carlezon WA Jr, Neve RL, Nestler EJ, Damis M: Long-term memory is facilitated by CREB overexpression in the amygdala. J Neurosci 2001; 21:2402–2412Google Scholar
153. Lenox RH, Gould TD, Manji HK: Endophenotypes in bipolar disorder. Am J Med Genet 2002; 114:391–406Crossref, Medline, Google Scholar
154. Tremblay LK, Naranjo CA, Cardenas L, Herrmann N, Busto UE: Probing brain reward system function in major depressive disorder: altered response to dextroamphetamine. Arch Gen Psychiatry 2002; 59:409–416Crossref, Medline, Google Scholar
155. Cardenas L, Tremblay LK, Naranjo CA, Herrmann N, Zack M, Busto UE: Brain reward system activity in major depression and comorbid nicotine dependence. J Pharmacol Exp Ther 2002; 302:1265–1271Crossref, Medline, Google Scholar
156. Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE: Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 2001; 8:229–242Crossref, Medline, Google Scholar
157. LeDoux JE: Emotion circuits in the brain. Annu Rev Neurosci 2000; 23:155–184Crossref, Medline, Google Scholar
158. Schafe GE, Nader K, Blair HT, LeDoux JE: Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 2001; 24:540–546Crossref, Medline, Google Scholar
159. Shumyatsky G, Tsvetkov E, Malleret G, Vronskaya S, Horton M, Hampton L, Battey JF, Dulac C, Kandel ER, Bolshakov VY: Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 2002; 111:905–918Crossref, Medline, Google Scholar
160. Walker DL, Davis M: Involvement of NMDA receptors within the amygdala in short versus long-term memory for fear conditioning as assessed with fear-potentiated startle. Behav Neurosci 2000; 114:1019–1033Crossref, Medline, Google Scholar
161. Rodrigues SM, Schafe GE, LeDoux JE: Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci 2001; 21:6889–6896Crossref, Medline, Google Scholar
162. Grillon C: Associative learning deficits increase symptoms of anxiety in humans. Biol Psychiatry 2002; 51:851–858Crossref, Medline, Google Scholar
163. Bauer EP, Schafe GE, LeDoux JE: NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 2002; 22:5239–5249Crossref, Medline, Google Scholar
164. McGaugh JL: Memory consolidation and the amygdala: a systems perspective. Trends Neurosci 2002; 25:456–461Crossref, Medline, Google Scholar
165. Killcross S, Robbins TW, Everitt BJ: Different types of fear-conditioned behavior mediated by separate nuclei within amygdala. Nature 1997; 388:377–380Crossref, Medline, Google Scholar
166. Wilensky AE, Schafe GE, LeDoux JE: The amygdala modulates memory consolidation of fear-motivated inhibitory avoidance learning but not classical fear conditioning. J Neurosci 2000; 20:7059–7066Crossref, Medline, Google Scholar
167. McIntyre CK, Hatfield T, McGaugh JL: Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats. Eur J Neurosci 2002; 16:1223–1226Crossref, Medline, Google Scholar
168. McGaugh JL, Roozendaal B: Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol 2002; 12:205–210Crossref, Medline, Google Scholar
169. Roozendaal B, Brunson KL, Holloway BL, McGaugh JL, Baram TZ: Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc Natl Acad Sci USA 2002; 99:13908–13913Crossref, Medline, Google Scholar
170. Roesler R, Roozendaal B, McGaugh JL: Basolateral amygdala lesions block the memory-enhancing effect of 8-Br-cAMP infused into the entorhinal cortex of rats after training. Eur J Neurosci 2002; 15:905–910Crossref, Medline, Google Scholar
171. Debiec J, LeDoux JE, Nader K: Cellular and systems reconsolidation in the hippocampus. Neuron 2002; 36:527–538Crossref, Medline, Google Scholar
172. Milekic MH, Alberini CM: Temporally graded requirement for protein synthesis following memory reactivation. Neuron 2002; 36:521–525Crossref, Medline, Google Scholar
173. Myers KM, Davis M: Systems-level reconsolidation: re-engagement of the hippocampus with memory reactivation. Neuron 2002; 36:340–343Crossref, Medline, Google Scholar
174. Przybyslawski J, Roullet P, Sara SJ: Attenuation of emotional and nonemotional memories after their reactivation: role of β adrenergic receptors. J Neurosci 1999; 19:6623–6628Crossref, Medline, Google Scholar
175. Sara SJ: Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 2000; 7:73–84Crossref, Medline, Google Scholar
176. Nader K, Schafe GE, LeDoux JE: Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 2000; 406:722–726Crossref, Medline, Google Scholar
177. Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ: CREB required for the stability of new and reactivated fear memories. Nat Neurosci 2002; 4:348–355Crossref, Google Scholar
178. Przybyslawski J, Sara SJ: Reconsolidation of memory after its reactivation. Behav Brain Res 1997; 84:241–246Crossref, Medline, Google Scholar
179. Myers KM, Davis M: Behavioral and neural analysis of extinction. Neuron 2002; 36:567–584Crossref, Medline, Google Scholar
180. Cain CK, Blouin AM, Barad MG: L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci 2002; 22:9113–9121Crossref, Medline, Google Scholar
181. McGaugh JL, Castellano C, Brioni J: Picrotoxin enhances latent extinction of conditioned fear. Behav Neurosci 1990; 104:264–267Crossref, Medline, Google Scholar
182. Willick ML, Kokkinidis L: Cocaine enhances the expression of fear-potentiated startle: evaluation of state-dependent extinction and the shock-sensitization of acoustic startle. Behav Neurosci 1995; 109:929–938Crossref, Medline, Google Scholar
183. Quirk GJ, Russo GK, Barron JL, Lebron K: The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 2000; 20:6225–6231Crossref, Medline, Google Scholar
184. Morgan MA, Romanski LM, LeDoux JE: Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett 1993; 163:109–113Crossref, Medline, Google Scholar
185. Milad MR, Quirk GJ: Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 2002; 420:70–74Crossref, Medline, Google Scholar
186. Royer S, Martina M, Pare D: Bistable behavior of inhibitory neurons controlling impulse traffic through the amygdala: role of a slowly deinactivating K+ current. J Neurosci 2000; 20:9034–9039Crossref, Medline, Google Scholar
187. Herry C, Garcia R: Prefrontal cortex long-term potentiation but not long-term depression is associated with maintenance of extinction of learned fear in mice. J Neurosci 2002; 22:577–583Crossref, Medline, Google Scholar
188. Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS: Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry 1999; 45:806–816Crossref, Medline, Google Scholar
189. Davis M: The role of NMDA receptors and MAP kinase in the amygdala in extinction of fear: clinical implications for exposure therapy. Eur J Neurosci 2002; 16:395–398Crossref, Medline, Google Scholar
190. Young LJ: The neurobiology of social recognition, approach, and avoidance. Biol Psychiatry 2002; 51:18–26Crossref, Medline, Google Scholar
191. Ferguson JN, Young LJ, Hearn EF, Insel TR, Winslow JT: Social amnesia in mice lacking the oxytocin gene. Nat Genet 2000; 25:284–288Crossref, Medline, Google Scholar
192. Ferguson JN, Aldag JM, Insel TR, Young LJ: Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci 2001; 21:8278–8285Crossref, Medline, Google Scholar
193. Carter CS, Getz LL: Monogamy and the prairie vole. Sci Am 1993; 268:100–106Crossref, Medline, Google Scholar
194. Shapiro LE, Dewsbury DA: Differences in affiliative behavior, pair bonding, and vaginal cytology in two species of vole (Microtus Ochrogaster and M montanus). J Comp Psychol 1990; 104:268–274Crossref, Medline, Google Scholar
195. Insel TR, Wang Z, Ferris CF: Patterns of brain vasopressin receptor distribution associated with social organization in microtine rodents. J Neurosci 1994; 14:5381–5392Crossref, Medline, Google Scholar
196. Young LJ, Winslow JT, Nilsen R, Insel TR: Species differences in V1a receptor gene expression in monogamous and non-monogamous voles: behavioral consequences. Behav Neurosci 1997; 111:599–605Crossref, Medline, Google Scholar
197. McBride WJ, Murphy JM, Ikemoto S: Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 1999; 101:129–152Crossref, Medline, Google Scholar
198. Rilling JK, Gutman DA, Zeh TR, Pagnoni G, Berns GS, Kilts CD: A neural basis for social cooperation. Neuron 2002; 35:395–405Crossref, Medline, Google Scholar
199. Dolan RJ: Emotion, cognition, and behavior. Science 2002; 298:1191–1194Crossref, Medline, Google Scholar
200. True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ, Nowak J: A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch Gen Psychiatry 1993; 50:257–264Crossref, Medline, Google Scholar
201. Comings DE, Muhleman D, Gysin R: Dopamine D2 receptor (DRD2) gene and susceptibility to posttraumatic stress disorder: a study and replication. Biol Psychiatry 1996; 40:368–372Crossref, Medline, Google Scholar
202. Gelernter J, Southwick S, Goodson S, Morgan A, Nagy L, Charney DS: No association between D2 dopamine receptor (DRD2) “A” system alleles or DRD2 haplotypes, and posttraumatic stress disorder. Biol Psychiatry 1999; 45:620–625Crossref, Medline, Google Scholar
203. Segman RH, Cooper-Kazaz R, Macciardi F, Goltser T, Halfon Y, Dobroborski T, Shalev AY: Association between the dopamine transporter gene and posttraumatic stress disorder. Mol Psychiatry 2002; 7:903–907Crossref, Medline, Google Scholar
204. Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, Delaney RC, McCarthy G, Charney DS, Innis RB: MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995; 152:973–981Link, Google Scholar
205. Bremner JD, Randall P, Vermetten E, Staib L, Bronen RA, Mazure C, Capelli S, McCarthy G, Innis RB, Charney DS: Magnetic resonance imaging-based measurements of hippocampal volume in posttraumatic stress disorder related to childhood physical and sexual abuse—a preliminary report. Biol Psychiatry 1997; 41:23–32Crossref, Medline, Google Scholar
206. Gurvits TV, Shenton ME, Hokama H, Ohta H, Lasko NB, Gilbertson MW, Orr SP, Kikinis R, Jolesz FA, McCarley RW, Pitman RK: Magnetic resonance imaging study of hippocampal volume in chronic, combat-related posttraumatic stress disorder. Biol Psychiatry 1996; 40:1091–1099Crossref, Medline, Google Scholar
207. Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, Orr SP, Pitman RK: Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 2002, 5:1242–1247Google Scholar
208. DeRijk RH, Schaaf M, de Kloet ER: Glucocorticoid receptor variants: clinical implications. J Steroid Biochem Mol Biol 2002; 81:103–122Crossref, Medline, Google Scholar
209. Small KM, Wagoner LE, Levin AM, Kardia SLR, Liggett ST: Synergistic polymorphisms of β1 and α2c adrenergic receptors and the risk of congestive heart failure. N Engl J Med 2002; 347:1135–1142Crossref, Medline, Google Scholar
210. Kallio J, Pesonen U, Kaipio K, Karvonen MK, Jaakkola U, Heinonen OJ, Uusitupa MI, Koulu M: Altered intracellular processing and release of neuropeptide Y due to leucine 7 to proline 7 polymorphism in the signal peptide of preproneuropeptide Y in humans. FASEB J 2001; 15:1242–1244Crossref, Medline, Google Scholar
211. Goddard AW, Mason GF, Rothman DL, Behar KL, Petroff OA, Charney DS, Krystal JH: Reductions in occipital cortex GABA levels in panic disorder. Arch Gen Psychiatry 2001; 58:556–561Crossref, Medline, Google Scholar
212. Ishikawa-Brush Y, Powell JF, Bolton P, Miller AP, Francis F, Willard HF, Lehrach H, Monaco AP: Autism and multiple exostoses associated with an X-8 translocation occurring within the GRPR gene and 3′ to the SDC2 gene. Hum Mol Genet 1997; 6:1241–1250Crossref, Medline, Google Scholar
213. Insel TR, Young LJ: The neurobiology of attachment. Nat Rev Neurosci 2001; 2:129–136Crossref, Medline, Google Scholar
214. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR: Serotonin transporter genetic variation and the response of the human amygdala. Science 2002; 297:400–403Crossref, Medline, Google Scholar
215. Garpenstrand H, Annas P, Ekblom J, Oreland L, Fredrikson M: Human fear conditioning is related to dopaminergic and serotonergic biological markers. Behav Neurosci 2001; 115:358–364Crossref, Medline, Google Scholar
216. Holmes A, Yang RJ, Murphy DL, Crawley JN: Evaluation of antidepressant-related behavioral responses to mice lacking the serotonin transporter. Neuropsychopharmacology 2002; 27:914–923Crossref, Medline, Google Scholar
217. Plato, vol II: Laches, Protogoras, Meno Euth Demus. Washington, DC, Howard University Press, 1924, pp 1–85Google Scholar
218. Rutter M: The interplay of nature, nurture, and developmental influences. Arch Gen Psychiatry 2002, 59:996–1000Google Scholar
219. Masten AS: Resilience processes in development. Am Psychol 2001; 227–238Google Scholar

