
Olanzapine Versus Haloperidol Treatment in First-Episode Psychosis
Abstract
OBJECTIVE: It has been hypothesized that the morbidity and mortality associated with schizophrenia can be prevented by providing effective treatment during the first episode of psychosis. Hence, the authors examined patients with first-episode psychosis to determine the efficacy and safety of olanzapine and haloperidol treatment. METHOD: A subpopulation of first-episode patients (N=83) from a large prospective, multicenter, international, double-blind, 6-week acute treatment study was evaluated. These patients were selected from a pool of 1,996 patients who had a DSM-III-R diagnosis of schizophrenia, schizoaffective disorder, or schizophreniform disorder and who also met the following criteria: 1) the length of their current psychotic episode had to be 5 or fewer years, and 2) patients had to be 45 years of age or younger at onset of first psychotic symptoms. RESULTS: Compared to haloperidol, olanzapine showed a statistically significantly greater reduction in the Brief Psychiatric Rating Scale (BPRS) total and negative scores and in the Positive and Negative Syndrome Scale total and positive scores. Clinical response (defined as 40% or greater improvement in BPRS total score from baseline) was also statistically significantly higher in olanzapine-treated patients (67.2%) than in haloperidol-treated patients (29.2%). Olanzapine-treated patients further showed statistically significant improvements in the Simpson-Angus scale and Barnes Akathisia Scale scores, while haloperidol-treated patients showed a worsening on both measures. Compared to olanzapine-treated multiple-episode patients in the parent study, olanzapine-treated first-episode patients achieved an even statistically significantly higher response. Haloperidol-treated first-episode patients experienced statistically significantly more extrapyramidal symptoms than haloperidol-treated multiple-episode patients. CONCLUSIONS: In patients experiencing first-episode psychosis, olanzapine had a risk-benefit profile significantly superior to that of haloperidol. The study results suggest that novel antipsychotic agents such as olanzapine should be considered as a preferred option in first-episode psychosis, on the basis of both safety and efficacy advantages.
Schizophrenia is a severe and disabling psychiatric disorder with devastating effects on both its victims and their families. Furthermore, it extracts enormous economic costs from society (1). With a typical onset in late adolescence or early adulthood, schizophrenia generally follows a recurrent and chronic course. While the majority of patients recover from their psychotic symptoms following their first episode of illness (2), they are subsequently at a high risk for a relapse and, ultimately, for persistent morbidity in the form of residual positive or negative symptoms or both, neurocognitive impairment, and deficits in social and vocational performance (3, 4). Moreover, the management of these patients is complicated by a high rate of adverse neurologic complications including parkinsonism, dystonia, akathisia, neuroleptic malignant syndrome, and tardive dyskinesia associated with conventional neuroleptic drugs (5, 6).
Longitudinal studies have demonstrated that much of the progression of schizophrenia occurs early, during the first 5 years following the first psychotic episode (7). Previous studies with first- and multiple-episode patients (8) suggest that this early progression may reflect an active pathophysiologic process that, if sustained, contributes to the enduring impairment associated with chronic schizophrenia (9–11). However, several longitudinal studies (12–14) have suggested that the cognitive deficits of schizophrenia may be stable or actually improve early in the course of the illness with treatment. This suggests that the cognitive pathology of schizophrenia may follow a different temporal and clinical course than the positive and negative symptom dimensions of the illness or that the methodological complexities of characterizing the longitudinal course of cognitive function cannot yet be fully resolved (15). Nevertheless, therapeutic intervention in the early phase of the illness has increasingly been recognized as of critical importance (11, 16).
Conventional neuroleptic agents have been the mainstay in the pharmacotherapy of schizophrenia. While they are effective in acute positive symptoms, there are serious limitations in their efficacy for other aspects of the illness, and they are associated with a high prevalence of side effects, rendering them a suboptimal treatment modality (17–21). Thus, it is not surprising that compliance rates with these conventional agents have been low. Poor compliance or noncompliance during the early stages of schizophrenia may complicate clinical outcomes. Furthermore, even effective maintenance treatment with conventional neuroleptic drugs may not prevent the progression of schizophrenia. Moreover, through iatrogenic effects, conventional neuroleptics may adversely affect patients’ abilities to function and develop normally.
With the emergence of novel antipsychotic drugs, expectations for a superior therapeutic profile have arisen. Scientific evidence indicates that clozapine, the first of these novel agents, has superior clinical efficacy in treatment-refractory and -intolerant patients with schizophrenia (22). However, it has not been conclusively determined whether clozapine has superior clinical efficacy in the treatment of nonrefractory schizophrenia and whether it can prevent the clinical progression of schizophrenia, that is, the psychopathologic, cognitive, functional, and neurologic deterioration. Nonetheless, there are compelling reasons to believe that this would be the case, on the basis of both preclinical and clinical observations (23–26). However, clozapine’s serious side effects have limited its application to treatment-refractory and -intolerant patient populations.
Unfortunately, there have been very few comparisons of novel to conventional antipsychotic drugs in first-episode patients, particularly through use of a double-blind, randomized design (27, 28).
Olanzapine, a thienobenzodiazepine, shares a similar chemical structure with clozapine, as well as broad-spectrum in vitro and animal neuropharmacology (29–32). On the basis of clinical studies conducted in its development, olanzapine has therapeutic efficacy that is at least comparable (and in some instances superior) to that of conventional antipsychotic drugs and has a low propensity to induce extrapyramidal symptoms (33–38).
On the basis of evidence derived from multiple-episode patients, we hypothesized that olanzapine would also demonstrate an efficacy and safety profile superior to that of haloperidol among patients experiencing a first episode of psychosis. To test this hypothesis, we examined a subpopulation of first-episode patients (N=83) participating in a multicenter clinical trial of olanzapine versus haloperidol involving a total of 1,996 randomized patients with schizophrenia, schizophreniform disorder, or schizoaffective disorder (39). The primary objective of the trial was to examine the overall clinical efficacy and safety of olanzapine; thus, research questions focusing on first-episode psychosis were not formulated in the design of the parent study (39). The hypothesis that olanzapine is more effective and safer than conventional antipsychotics in treating first-episode psychosis was developed after the analyses addressing the primary objectives of the parent trial were completed.
METHOD
The data for these analyses were taken from the 6-week acute phase of a large prospective, international, multicenter, double-blind study previously reported (39). After a 2–9-day screening and washout period, patients were randomly assigned to study drug in a 2:1 (olanzapine:haloperidol) ratio. All patients began therapy with 5 mg/day of study drug; after each 7-day period, the study drug could be adjusted in 5-mg increments or decrements within the allowed dose range of 5–20 mg/day. During the study, benztropine, up to 6 mg/day, could be prescribed to treat emergent extrapyramidal symptoms, but prophylactic use was discouraged. Limited use of benzodiazepines was also allowed as concomitant medication. The study was approved by each institution’s ethical review board, and signed informed consent forms were obtained from all patients before their participation.
Patients had to meet DSM-III-R criteria for schizophrenia, schizophreniform disorder, or schizoaffective disorder. Diagnoses were established by clinical interview, chart review, physical examination, and laboratory testing. In addition to the diagnosis, patients were required to be actively symptomatic, as evidenced by a minimum Brief Psychiatric Rating Scale (BPRS) total score of 18, or be intolerant of current antipsychotic therapy (excluding haloperidol), as evidenced by a recent adverse event of sufficient gravity to warrant a switch in therapy. A total of 1,996 patients were randomly assigned to treatment: 1,336 to olanzapine and 660 to haloperidol.
Three additional criteria were added to the entry criteria of the parent study described earlier: patients must have been in their first episode of psychosis, the length of the current psychotic episode must have been no more than 5 years, and the patient’s age at onset of the first psychotic episode must have been no more than 45 years.
Efficacy assessments included the Positive and Negative Syndrome Scale (40), the BPRS extracted from the Positive and Negative Syndrome Scale, and the Montgomery-�sberg Depression Rating Scale (41). All rating scales were administered at each scheduled visit, with the exception of the Montgomery-�sberg scale, which was administered at baseline (before random assignment to study drug) and at the last visit during the acute phase of the study.
Adverse event experiences were assessed at each visit by spontaneously reported events. In addition, treatment-associated extrapyramidal symptoms and akathisia were rated objectively with the Simpson-Angus Scale (42) and the Barnes Akathisia Scale (43).
All analyses were done on an intent-to-treat basis; i.e., all randomized patients were included in the analysis. Patients were included in the analysis of change if they had both a baseline and a postbaseline observation. One olanzapine patient had no postbaseline observation. Total scores from rating scales were derived from the individual items; if any single item was missing, the total score was treated as missing.
For all continuous efficacy and safety measures, a two-sample t test was used to assess differences in treatment effect between olanzapine and haloperidol treatment groups. In addition, patients were dichotomized as responders or nonresponders. The protocol established the primary efficacy analysis as the mean change from baseline to endpoint last observation carried forward in the BPRS total score. Fisher’s exact test (two-tailed) was used to analyze treatment effects for categorical efficacy and safety measures. Predictors of response, defined as percentage change from baseline in BPRS total score, were investigated with the use of stepwise linear regression.
All cited p values were two-tailed with a significance level of 0.05 as specified in the protocol, except for predictors of response, which used a significance level of 0.15 to assess exploratory associations. No adjustments in p values for the multiplicity of tests were made; therefore, the error rate was on a comparisonwise basis. Statistical Analysis Software (44) was used for all analyses.
RESULTS
A total of 83 patients experiencing their first episode of psychosis and satisfying the selection criteria were randomly assigned to treatment with olanzapine (N=59) or haloperidol (N=24). No statistically significant differences were found between patients in the two treatment groups with regard to any of the characteristics shown in table 1. The majority of patients were male, Caucasian, and in their late 20s. The average age at onset of the psychotic episode was about 27 years. The average length of the psychotic episode was about 13 months; 56 (67.5%) of the patients had a psychotic episode of 1 year or less and 10 (12.0%) of the patients had a psychotic episode of between 1 and 2 years.
Nearly twice as many olanzapine-treated patients (N=43 of 59, 72.9%) as haloperidol-treated patients (N=9 of 24, 37.5%) completed the acute phase of the study (Fisher’s exact p=0.005). One-third (N=8 of 24, 33.3%) of haloperidol-treated patients discontinued treatment because of lack of efficacy, compared to 13.6% (N=8 of 59) of olanzapine-treated patients (Fisher’s exact p=0.06). Furthermore, haloperidol-treated patients were almost 10 times more likely than their olanzapine counterparts to discontinue therapy because of an adverse event (Fisher’s exact p=0.02). One olanzapine-treated patient discontinued the acute phase because of an adverse event (depression). In contrast, four haloperidol-treated patients (16.7%) discontinued the acute phase because of an adverse event (akathisia, hypertonia, abnormal thinking, and extrapyramidal syndrome).
The modal dose for an individual patient was defined as the most frequently administered daily dose of study drug. The mean modal olanzapine and haloperidol doses, calculated for all first-episode patients, were 11.6 (SD=5.9) and 10.8 (SD=4.8) mg/day, respectively.
Efficacy
Endpoint analysis
Baseline scores and mean change from baseline to endpoint on the individual efficacy scales for the two treatment groups, along with 95% confidence intervals, are shown in table 2. No statistically significant differences in baseline severity were observed between the two treatment groups. At endpoint, olanzapine was statistically significantly superior to haloperidol in the reduction of BPRS total and negative scores and Positive and Negative Syndrome Scale total and positive scores.
Response rate analysis
A more robust measure of the respective treatment effects is a clinical response, defined a priori as a 40% or greater BPRS total improvement from baseline. Olanzapine-treated patients achieved a significantly higher response rate (N=39 of 58, 67.2%) than haloperidol-treated patients (N=7 of 24, 29.2%) (Fisher’s exact p=0.003). In addition, a significantly greater percentage of olanzapine-treated patients (N=48 of 58, 82.8%) than haloperidol-treated patients (N=14 of 24, 58.3%) achieved a reduction in BPRS total score of at least 20% from baseline (Fisher’s exact p=0.03).
Previous Neuroleptic Exposure
The previous neuroleptic exposure was determined for each patient on the basis of previous treatment history collected at the start of the study. The previous exposure for three patients could not be determined. The range of previous neuroleptic exposure varied widely. Of the 83 first-episode patients, 19 (23%) had no previous neuroleptic exposure (14 in the olanzapine group and five in the haloperidol group). The majority of the patients (N=49, 59%) had 2 months’ exposure or less to neuroleptics. Of the patients with 2 months’ exposure or less, 72% (N=26 of 36) of the olanzapine-treated patients responded, compared to 38% (N=5 of 13) of the haloperidol-treated patients (Fisher’s exact p=0.05). Of the patients with more than 2 months’ exposure to previous neuroleptics, 57% (N=12 of 21) of the olanzapine and 22% (N=2 of 9) of the haloperidol patients responded (Fisher’s exact p=0.12).
Predictors of Response
Several response predictors were investigated by using stepwise linear regression. These included therapy, age, age at onset, duration of current episode, gender, race, type of psychosis (schizophrenia, schizoaffective, schizophreniform), course of psychosis (schizophrenia: chronic, subchronic, or unspecified; schizoaffective: bipolar type currently depressed, bipolar type mixed, or depressive type currently depressed), and baseline severity on each efficacy scale, along with the interaction of therapy with gender, course, and age at onset. The percentage change from baseline in BPRS total score was used as the dependent variable. Therapy was the most important and only statistically significant predictor of response (R2=0.09, F=9.72, df=1, 77, p=0.003). The model predicted that olanzapine-treated patients would have a 25% greater decrease in BPRS total score than haloperidol-treated patients. The type of psychosis (R2=0.06, F=2.98, df=2, 77, p=0.06) and gender (R2=0.03, F=2.48, df=1, 77, p=0.12) were marginally significant predictors of response. Better response was associated more with men than with women (13% difference) and more with schizophreniform than with schizophrenic (9% difference) or schizoaffective (28% difference) patients.
Safety
Extrapyramidal symptoms
Extrapyramidal symptom ratings, including the Simpson-Angus scale (42) and Barnes Akathisia Scale (43), were analyzed to estimate the prevalence of extrapyramidal symptoms by both baseline-to-endpoint change and newly emergent categorical changes. The Simpson-Angus total score change from baseline to endpoint reflected an improvement in extrapyramidal symptoms among the olanzapine-treated patients (mean=–0.5, SD=3.3). In contrast, a worsening occurred among the haloperidol-treated patients (mean=4.5, SD=7.4). The difference of –5.0 points significantly favored olanzapine (t=4.15, df=79, p<0.001, 95% confidence interval=–7.3 to –2.6). A similar pattern emerged on the Barnes Akathisia Scale. Mean change on the Barnes global scores showed that olanzapine-treated patients improved from baseline (mean=–0.1, SD=0.8), whereas haloperidol-treated patients worsened from baseline (mean=0.5, SD=1.2). This treatment difference was statistically significant (t=2.90, df=79, p=0.005, 95% confidence interval=–1.1 to –0.2).
The percentage of patients with treatment-emergent parkinsonism (a total score higher than 3 on the Simpson-Angus scale at any postbaseline visit, given a total score of 3 or less at all baseline visits) was statistically significantly lower in the olanzapine than in the haloperidol treatment group (olanzapine: N=9 of 48, 18.8%; haloperidol: N=10 of 19, 52.6%) (Fisher’s exact p=0.01). Similarly, significantly fewer olanzapine-treated than haloperidol-treated patients experienced treatment-emergent akathisia (Barnes global score of 2 or more at any postbaseline visit, given a global score of less than 2 at all baseline visits) (olanzapine: N=6 of 53, 11.3%; haloperidol: N=8 of 21, 38.1%) (Fisher’s exact p=0.02).
Concomitant medication use
A significantly lower proportion of olanzapine-treated than haloperidol-treated patients took at least one dose of anticholinergic medication (olanzapine: N=8 of 59, 13.6%; haloperidol: N=10 of 24, 41.7%) (Fisher’s exact p=0.008). The mean daily dose of anticholinergic medication, expressed in benztropine equivalents, was 0.20 mg/day for olanzapine-treated patients and 0.93 mg/day for haloperidol-treated patients (t=1.96, df=81, p=0.05). The proportion of patients receiving at least one dose of benzodiazepine was similar for both treatment groups (olanzapine: N=30 of 59, 50.8%; haloperidol: N=16 of 24, 66.7%) (Fisher’s exact p=0.23).
Adverse events
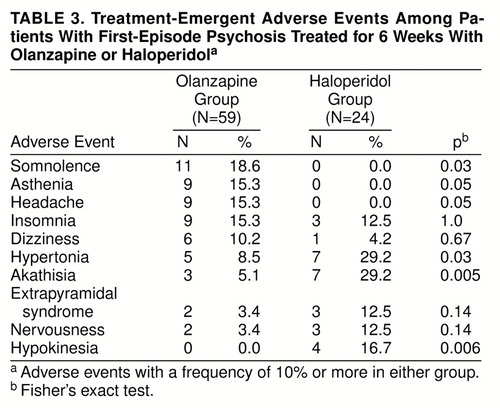
Table 3 shows treatment-emergent adverse events that occurred at a rate of at least 10% in either treatment group. Olanzapine-treated patients experienced a statistically significantly higher prevalence of treatment-emergent somnolence than haloperidol-treated patients, while haloperidol-treated patients experienced a statistically significantly greater prevalence of treatment-emergent akathisia, hypertonia, and hypokinesia than olanzapine-treated patients.
Vital signs, weight, and laboratory analyses
Assessments of vital signs revealed no clinically significant treatment differences. Statistically significantly more weight gain was observed in olanzapine-treated patients (4.1 kg) than haloperidol-treated patients (0.5 kg) (t=4.65, df=79, p<0.001).
Prolactin elevations were 4.5 times higher, on average, in the haloperidol treatment group at endpoint (t=4.85, df=62, p<0.001). Neither compound showed evidence of hematoxicity. Olanzapine treatment was associated with early, transient increases in alanine aminotransferase/serum glutamic-pyruvic transaminase in 16.1% (N=9 of 56) of patients; however, these elevations were not associated with clinical symptoms, were modest in scope, and did not necessitate the discontinuation of olanzapine. No other clinically relevant differences were evident.
Comparison of First-Episode and Multiple-Episode Patients
Of the original 1,996 patients randomized in the parent study, 83 patients experiencing their first episode of psychosis satisfied the selection criteria. These 83 patients were compared with the remaining 1,913 multiple-episode patients in the study who did not satisfy the selection criteria. In addition, within the olanzapine treatment group, the 59 first-episode patients were compared with the 1,277 remaining patients. Similarly, within the haloperidol treatment group, the 24 first-episode patients were compared with the 636 remaining patients.
Within each treatment group, the first-episode patients were, on average, 10.5 years younger, had their onset of first acute psychotic episode 3.4 years later, and had a duration of current psychotic episode an average of 2.0 years less than the multiple-episode patients. However, the two groups were similar in gender and race. At baseline, first-episode patients had statistically significantly lower BPRS negative scores (6.0 versus 6.7; t=1.98, df=1946, p=0.05), Positive and Negative Syndrome Scale negative scores (22.4 versus 24.2; t=2.41, df=1946, p=0.02), Simpson-Angus total scores (1.5 versus 2.8; t=2.64, df=1908, p=0.008), and Barnes akathisia scores (0.3 versus 0.6; t=2.37, df=1943, p=0.02) than multiple-episode patients.
The mean modal olanzapine and haloperidol doses, calculated for all multiple-episode patients, were 12.7 (SD=6.0) and 11.0 (SD=5.8) mg/day, respectively. In comparison, for first-episode patients the mean modal olanzapine and haloperidol doses were 11.6 (SD=5.9) and 10.8 (SD=4.8) mg/day, respectively.
Of those patients with a postbaseline measure, the 58 olanzapine first-episode patients had a response rate (40% or more improvement in BPRS total) of 67.2%, which was statistically significantly greater than the 45.1% response rate for the remaining 1,255 olanzapine multiple-episode patients (Fisher’s exact p=0.001). The 24 haloperidol first-episode patients had a response rate of 29.2%, which was similar to the 30.1% response rate seen in the 611 multiple-episode haloperidol patients (Fisher’s exact p=1.00). The comparison of first-episode and multiple-episode baseline to endpoint change in efficacy and extrapyramidal symptom scores for each treatment is given in table 4. There was statistically significantly greater mean improvement from baseline in the first-episode group than the multiple-episode group in BPRS total and positive scores and Positive and Negative Syndrome Scale total and positive scores for olanzapine-treated patients and in BPRS positive scores for haloperidol-treated patients. For haloperidol treatment, first-episode patients had a statistically significantly greater increase than multiple-episode patients in Simpson-Angus total scores from baseline to endpoint.
For haloperidol-treated patients, the following treatment-emergent adverse events with a frequency of 10% or more occurred statistically significantly more frequently in the first-episode patients (N=24) than the multiple-episode patients (N=635): hypertonia (29.2% versus 8.5%) (Fisher’s exact p=0.004) and hypokinesia (16.7% versus 0.5%) (Fisher’s exact p<0.001). For olanzapine-treated patients, asthenia was the only treatment-emergent adverse event with a frequency of 10% or more that occurred statistically significantly more frequently (15.3% versus 5.9%) (Fisher’s exact p=0.009) in first-episode patients (N=59) than in multiple-episode patients (N=1,278). No treatment-emergent adverse events occurred statistically significantly more frequently in multiple-episode patients than in first-episode patients for either treatment group.
DISCUSSION
The original hypothesis of these analyses was that olanzapine would demonstrate an efficacy and safety profile superior to that of haloperidol in the treatment of patients experiencing their first episode of psychosis. Our findings confirmed that olanzapine does, indeed, have several advantages over haloperidol in this population.
It is important in first-episode psychosis to introduce effective treatment as early as possible after the initial onset of psychotic symptoms. Studies have suggested that the longer the psychosis remains untreated or ineffectively treated, the worse the final prognosis may be (8, 11). In this report, olanzapine demonstrated a statistically significant superior reduction in BPRS total (primary efficacy outcome) and negative scores and Positive and Negative Syndrome Scale total and positive scores from baseline to endpoint, compared to haloperidol. Olanzapine-treated patients were over two times more likely to achieve a clinical response (defined as a 40% or greater improvement from baseline in BPRS total score) than haloperidol-treated patients (67.2% versus 29.2%). In support of this observation, a significantly higher percentage of olanzapine-treated patients (82.8%) than haloperidol-treated patients (58.3%) had a 20% or greater improvement from baseline in BPRS total score.
The majority of patients (59%) had 2 months or less of previous exposure to neuroleptics, suggesting that the first-episode group in this study was not primarily treatment resistant. For those patients with less than 2 months’ exposure, over 1.9 times as many olanzapine- as haloperidol-treated patients responded. For those patients with more than 2 months’ exposure, 2.6 times as many olanzapine-treated patients as haloperidol-treated patients responded. As expected, in both treatment groups the response rate was lower among patients with more than 2 months’ exposure to neuroleptic drugs than among those with 2 months’ or less exposure.
Predictors of clinical response to treatment with olanzapine or haloperidol were investigated. Therapy was the most important and only statistically significant predictor of response. Marginally statistically significant predictors of response were the type of psychosis and gender. The fact that schizophreniform patients performed better than schizophrenic or schizoaffective patients could be associated with the length of time that patients displayed active psychosis. According to DSM-III-R criteria, schizophreniform patients must have an episode of psychosis of 6 months or less in length, while schizophrenic and schizoaffective patients would typically have been suffering from psychotic illness for substantially longer periods of time. Therefore, this result is consistent with reports from other investigators (11, 45) showing that the longer the duration of illness, the worse the outcome. Female subjects typically respond better than male subjects to treatment for first-episode psychosis (2, 46); however, our analysis associated better response with men than with women. This result was probably driven by the fact that the female haloperidol patients did much worse than their male counterparts (mean changes in BPRS total scores: –4.4 versus –10.7). On the other hand, female and male olanzapine patients performed similarly (mean changes in BPRS total scores: –15.3 versus –17.1). When just the olanzapine patients were analyzed, gender was no longer an important predictor of response.
Olanzapine, when compared to haloperidol, demonstrated a superior safety profile. This may be especially important for first-episode patients in whom minimization of possible adverse events may have a decidedly positive effect on compliance. First-episode patients have been shown to be more sensitive to antipsychotic drugs, especially to extrapyramidal symptoms (47). For example, approximately 70%–80% of neuroleptic-naive patients typically develop side effects, especially parkinsonism (48), on exposure to a neuroleptic.
If the goal of treatment is adequate treatment duration, then noncompliance may ultimately adversely affect the outcome of the disease (8, 11). In the present study, the number of patients discontinuing treatment because of an adverse event was statistically significantly less with olanzapine (N=1) than with haloperidol (N=4, 16.6%). Moreover, haloperidol-treated patients experienced higher rates of treatment-emergent akathisia, hypertonia, and hypokinesia. Olanzapine-treated patients experienced only a greater rate of treatment-emergent somnolence.
Objective assessment of extrapyramidal symptoms, with the Simpson-Angus Scale (42) and the Barnes Akathisia Scale (43), clearly demonstrated that olanzapine-treated patients had a statistically significant improvement from baseline, whereas haloperidol-treated patients had a significant worsening. Treatment-emergent akathisia and parkinsonism were also statistically significantly less frequent in the olanzapine than in the haloperidol treatment group. In addition, a significantly smaller proportion of olanzapine- than haloperidol-treated patients took at least one dose of anticholinergic medication, and the mean daily dose of anticholinergic medication for olanzapine-treated patients was almost one-fifth that of the haloperidol-treated patients. These data from first-episode patients seem to confirm the lower extrapyramidal symptom potential of olanzapine that was found in other trials involving more chronically ill individuals (33, 34).
Olanzapine-treated patients had greater weight gain than haloperidol-treated patients. All of the novel antipsychotics recently released appear to share this adverse event. Both clozapine (49–51) and risperidone (52, 53) demonstrate significantly more weight gain in patients than haloperidol or typical antipsychotics.
Prolactin increases were higher in the haloperidol treatment group than in the olanzapine treatment group. Hyperprolactinemia may lead to a number of clinical sequelae (e.g., galactorrhea, amenorrhea, irregular menses, anovulation, impotence, azoospermia, gynecomastia) that can require a dose reduction (which may result in inadequate therapy) or even discontinuation of the neuroleptic agent (54).
The parent study involved 1,996 patients, of whom 83 patients were determined to be experiencing their first episode of psychosis. Not surprisingly, first-episode patients were generally younger than multiple-episode patients and had a shorter duration of the current episode. In addition, first-episode patients had lower mean extrapyramidal symptoms and negative symptom scores at baseline than multiple-episode patients, probably reflecting differences in their previous exposure to neuroleptics. The mean modal olanzapine dose used with first-episode patients was slightly lower than that used in multiple-episode patients; the mean modal haloperidol dose did not differ between first- and multiple-episode patients.
First-episode patients treated with olanzapine were significantly more likely to achieve a clinical response than multiple-episode patients treated with olanzapine (67.2% versus 45.1%). In addition, olanzapine-treated first-episode patients had statistically significantly greater mean improvement in BPRS total and positive scores and Positive and Negative Syndrome Scale total and positive scores than olanzapine-treated multiple-episode patients. In contrast, haloperidol had a similar response rate in both first-episode (29.3%) and multiple-episode (30.1%) patients. These results have significant potential relevance in antipsychotic drug selection for first-episode patients. Whereas the haloperidol treatment demonstrated lower overall response rates, which were comparable in both first- and multiple-episode patients, olanzapine treatment appeared to bestow even greater effects in subjects early in the course of their illness.
As measured by the Barnes and Simpson-Angus scale scores, first-episode haloperidol-treated patients experienced more extrapyramidal symptoms than multiple-episode patients, while there was no difference in extrapyramidal symptoms in either group of olanzapine-treated patients. In addition, the treatment-emergent extrapyramidal symptoms of hypertonia and hypokinesia occurred statistically significantly more frequently in haloperidol-treated first-episode patients than in haloperidol-treated multiple-episode patients. Only treatment-emergent asthenia occurred statistically significantly more frequently in olanzapine-treated first-episode than in olanzapine-treated multiple-episode patients. The results suggest that there were also clinically meaningful safety advantages with olanzapine in both multiple-episode and first-episode patients.
The primary limitation to these results is that they represent a post hoc stratification of the parent study. In addition, the group size was small. The primary strength of these results is that they come from a double-blind, well-controlled clinical trial comparing a novel antipsychotic with a conventional neuroleptic. Very few studies of this rigorous design are available.
In summary, for patients experiencing their first episode of psychosis, olanzapine showed superior treatment effectiveness, consisting of both safety and efficacy advantages, when compared to the conventional neuroleptic haloperidol. In a patient population in which early and effective treatment may attenuate some of the long-term effects of schizophrenia, it would seem prudent to select the antipsychotic agent with the optimal risk-benefit profile and greatest likelihood for sustained compliance. Further studies to corroborate the treatment advantages seen with olanzapine in first-episode psychotic patients are awaited.
Received Dec. 19, 1997; revision received May 22, 1998; accepted June 8, 1998. From Lilly Research Laboratories, Eli Lilly and Company, Lilly Corporate Center; Department of Psychiatry, University of North Carolina School of Medicine, Chapel Hill; and Department of Psychiatry, Harvard Medical School, and Department of Epidemiology, Harvard School of Public Health, Boston. Address reprint requests to Dr. Sanger, Lilly Research Laboratories, Eli Lilly and Company, Lilly Corporate Center, Drop Code 0537, Indianapolis, IN 46285; [email protected] (e-mail). Sponsored by Eli Lilly and Company.
 |
 |
 |
 |
1. Torrey EF (ed): Surviving Schizophrenia: A Manual for Families, Consumers, and Providers. New York, HarperCollins, 1995, p 409Google Scholar
2. Tohen M, Stoll AL, Strakowski SM, Faedda GL, Mayer PV, Goodwin DC, Kolbrener ML, Madigan AM: The McLean First-Episode Psychosis Project: six-month recovery and recurrence outcome. Schizophr Bull 1992; 18:273–282Crossref, Medline, Google Scholar
3. Ciompi L: The natural history of schizophrenia in the long term. Br J Psychiatry 1980; 136:412–420Crossref, Google Scholar
4. Hegarty JD, Baldessarini RJ, Tohen M, Waternaux C, Oepen G: One hundred years of schizophrenia: a meta-analysis of the outcome literature. Am J Psychiatry 1994; 151:1409–1416Link, Google Scholar
5. Chakos MH, Lieberman JA, Bilder RM, Borenstein M, Lerner G, Bogerts B, Wu H, Kinon B, Ashtari M: Increase in caudate nuclei volumes of first-episode schizophrenic patients taking antipsychotic drugs. Am J Psychiatry 1994; 151:1430–1436Link, Google Scholar
6. Levinson DF, Simpson GM, Singh H, Yadalam K, Jain A, Stephanos MJ, Silver P: Fluphenazine dose, clinical response, and extrapyramidal symptoms during acute treatment. Arch Gen Psychiatry 1990; 47:761–768Crossref, Medline, Google Scholar
7. McGlashan TH: A selective review of recent North American long-term follow-up studies of schizophrenia. Schizophr Bull 1988; 14:515–542Crossref, Medline, Google Scholar
8. Wyatt RJ: Neuroleptics and the natural course of schizophrenia. Schizophr Bull 1991; 17:325–351Crossref, Medline, Google Scholar
9. Lieberman JA, Kinon BJ, Loebel AD: Dopaminergic mechanisms of idiopathic and drug induced psychosis. Schizophr Bull 1990; 16:97–110Crossref, Medline, Google Scholar
10. Lieberman JA, Alvir JM, Koreen A, Geisler S, Chakos M, Sheitman B, Woerner M: Psychobiologic correlates of treatment response in schizophrenia. Neuropsychopharmacology 1996; 14(March suppl):13S–21SGoogle Scholar
11. Loebel AD, Lieberman JA, Alvir JMJ, Mayerhoff DI, Geisler SH, Szymanski SR: Duration of psychosis and outcome in first-episode schizophrenia. Am J Psychiatry 1992; 149:1183–1188Link, Google Scholar
12. Hoff AL, Riordan H, O’Donnell D, Stritzke P, Neale C, Boccio A, Anand AK, DeLisi LE: Anomalous lateral sulcus asymmetry and cognitive function in first-episode schizophrenia. Schizophr Bull 1992; 18:257–272; correction, 1994; 20:248Google Scholar
13. Nopoulos P, Flashman L, Flaum M, Arndt S, Andreasen N: Stability of cognitive functioning early in the course of schizophrenia. Schizophr Res 1994; 14:29–37Crossref, Medline, Google Scholar
14. Sweeney JA, Haas GL, Keilp JG, Long M: Evaluation of the stability of neuropsychological functioning after acute episodes of schizophrenia: one-year follow-up study. Psychiatry Res 1991; 38:63–76Crossref, Medline, Google Scholar
15. Keefe RSE, Perkins D, Lieberman JA: The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: a meta-analysis and methodological review. Schizophr Bull 1998 (in press)Google Scholar
16. Wyatt RJ: Early intervention for schizophrenia: can the course of the illness be altered? Biol Psychiatry 1995; 38:1–3Google Scholar
17. Lieberman JA: Understanding the mechanism of atypical antipsychotic drugs: a review of compounds in use and development. Br J Psychiatry 1993; 163(suppl 22):7–18Google Scholar
18. Gerlach J: New antipsychotics: classification, efficacy and adverse effects. Schizophr Bull 1991; 17:298–309Crossref, Google Scholar
19. Cassens G, Inglis AK, Appelbaum PS, Gutheil TG: Neuroleptics: effects on neuropsychological function in chronic schizophrenic patients. Schizophr Bull 1990; 16:717–725Crossref, Google Scholar
20. Hoff AL, Riordan H, O’Donnell DW, Morris L, DeLisi LE: Neuropsychological functioning of first-episode schizophreniform patients. Am J Psychiatry 1992; 149:898–903Link, Google Scholar
21. Medalia A, Gold JM, Merriam A: The effects of neuroleptics on neuropsychological test results of schizophrenics. Arch Clin Neuropsychol 1988; 3:249–271Crossref, Medline, Google Scholar
22. Safferman A, Lieberman JA, Kane J, Szymanski SR, Kinon B: Update on the clinical efficacy and side effects of clozapine. Schizophr Bull 1991; 17:247–261Crossref, Medline, Google Scholar
23. Green AI, Schildkraut JJ: Should clozapine be a first-line treatment for schizophrenia? the rationale for a double-blind clinical trial in first-episode patients. Harvard Rev Psychiatry 1995; 3:1–9Crossref, Medline, Google Scholar
24. Lipska BJ, Jaskiw GE, Braun AR, Weinberger DR: Prefrontal cortical and hippocampal modulation of haloperidol-induced catalepsy and apomorphine-induced stereotypic behaviors in the rat. Biol Psychiatry 1995; 38:255–262Crossref, Medline, Google Scholar
25. Olney JW, Farber NB: Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 1995; 52:998–1007Crossref, Medline, Google Scholar
26. Lieberman JA, Sheitman B, Kinon BJ: Neurochemical sensitization in the pathophysiology of schizophrenia: deficits and dysfunction in neuronal regulation and plasticity. Neuropsychopharmacology 1997; 17:205–229Crossref, Medline, Google Scholar
27. Emsley RA, McCreadie RG, Livingston M, DeSmedt G, Lemmens P: Risperidone in the treatment of first-episode patients with schizophreniform disorder: a double-blind multicenter study (abstract). J Eur Neuropsychopharmacology 1995; 5(3):350Google Scholar
28. Robinson DG, Lieberman JA, Sheitman B, Alvir JM, Kane JM: Pilot study of atypical antipsychotic agents in first episode schizophrenia. Schizophr Res 1997; 24:196Crossref, Google Scholar
29. Seeman P, Van Tol HH: Dopamine receptor pharmacology. Curr Opin Neurol Neurosurg 1993; 6:602–608Medline, Google Scholar
30. Bymaster FP, Calligaro DO, Falcone JF, Marsh RD, Moore NA, Tye NS, Seeman P, Wong DT: Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996; 14:87–96Crossref, Medline, Google Scholar
31. Moore NA, Calligaro DO, Wong DT, Bymaster FP, Tye NC: The pharmacology of olanzapine and other new antipsychotic agents. Curr Opin Invest Drugs 1993; 2:281–293Google Scholar
32. Moore NA, Tye NC, Axton MS, Risius FC: The behavioral pharmacology of olanzapine, a novel “atypical” antipsychotic agent. J Pharmacol Exp Ther 1992; 262:545–551Medline, Google Scholar
33. Beasley CM, Sanger T, Satterlee W, Tollefson G, Tran P, Hamilton S, Olanzapine HGAP Study Group: Olanzapine versus placebo: results of a double-blind, fixed-dose olanzapine trial. Psychopharmacology (Berl) 1996; 124:159–167Crossref, Medline, Google Scholar
34. Beasley CM, Tollefson G, Tran P, Satterlee W, Sanger T, Hamilton S, Olanzapine HGAP Study Group: Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology 1996; 14:105–118Crossref, Medline, Google Scholar
35. Beasley CM, Hamilton S, Crawford AM, Dellva MA, Tollefson G, Tran P, Blin O, Beuzen JN, Olanzapine E003 Study Group: Olanzapine versus haloperidol: acute-phase results of the international double-blind olanzapine trial. Eur Neuropsychopharmacol 1997; 7:125–137Crossref, Medline, Google Scholar
36. Tollefson GD, Beasley CM Jr, Tamura RN, Tran PV, Potvin JH: Blind, controlled, long-term study of the comparative incidence of treatment-emergent tardive dyskinesia with olanzapine or haloperidol. Am J Psychiatry 1997; 154:1248–1254Link, Google Scholar
37. Tran P, Dellva MA, Tollefson G, Beasley CM, Potvin JH, Kiesler GM: Extrapyramidal symptoms and tolerability of olanzapine versus haloperidol in the acute treatment of schizophrenia. J Clin Psychiatry 1997; 58:205–211Crossref, Medline, Google Scholar
38. Tollefson GD, Sanger TM: Negative symptoms: a path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am J Psychiatry 1997; 154:466–474Link, Google Scholar
39. Tollefson GD, Beasley CM Jr, Tran PV, Street JS, Krueger JA, Tamura RN, Graffeo KA, Thieme ME: Olanzapine versus haloperidol in the treatment of schizophrenia and schizoaffective and schizophreniform disorders: results of an international collaborative trial. Am J Psychiatry 1997; 154:457–465Link, Google Scholar
40. Kay SR, Fiszbein A, Opler LA: The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276Crossref, Medline, Google Scholar
41. Montgomery SA, �sberg M: A new depression scale designed to be sensitive to change. Br J Psychiatry 1979; 134:382–389Crossref, Medline, Google Scholar
42. Simpson GM, Angus JWS: A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 1970; 212:11–19Crossref, Medline, Google Scholar
43. Barnes TR: A rating scale for drug-induced akathisia. Br J Psychiatry 1989; 154:672–676Crossref, Medline, Google Scholar
44. SAS/STAT User"s Guide, version 6, 4th ed, vols 1, 2. Cary, NC, SAS Institute, 1990Google Scholar
45. Larsen TK, McGlashan TH, Johannessen JO, Vibe-Hansen L: First-episode schizophrenia, II: premorbid patterns by gender. Schizophr Bull 1996; 22:257–269Crossref, Medline, Google Scholar
46. Szymanski S, Lieberman JA, Alvir JM, Mayerhoff D, Loebel AD, Geisler S, Chakos M, Koreen A, Jody D, Kane J, Woerner M, Cooper T: Gender differences in onset of illness, treatment response, course, and biologic indexes in first-episode schizophrenic patients. Am J Psychiatry 1995; 152:698–703Link, Google Scholar
47. McEvoy J: Dosing issues: target symptoms and dose response. Psychiatr Annals 1995; 25:297–300Crossref, Google Scholar
48. McCreadie RG: Managing the first episode of schizophrenia: the role of new therapies. Eur Neuropsychopharmacol 1996; 6(suppl 2):S3–S5Google Scholar
49. Hummer M, Kemmler G, Kurz M, Kurzthaler I, Oberbauer H, Fleischhacker WW: Weight gain induced by clozapine. Eur Neuropsychopharmacol 1995; 5:437–440Crossref, Medline, Google Scholar
50. Buchanan RW: Clozapine: efficacy and safety. Schizophr Bull 1995; 21:579–591Crossref, Medline, Google Scholar
51. Lamberti JS, Bellnier T, Schwarzkopf SB: Weight gain among schizophrenic patients treated with clozapine. Am J Psychiatry 1992; 149:689–690Link, Google Scholar
52. Umbricht D, Kane JM: Medical complications of new antipsychotic drugs. Schizophr Bull 1996; 22:475–483Crossref, Medline, Google Scholar
53. Owens DG: Extrapyramidal side effects and tolerability of risperidone: a review. J Clin Psychiatry 1994; 55(5 suppl):29–35Google Scholar
54. Marken PA, Haykal RF, Fisher JN: Management of psychotropic-induced hyperprolactinemia. Clin Pharm 1992; 11:851–856Medline, Google Scholar

