
Association of Methylphenidate-Induced Craving With Changes in Right Striato-orbitofrontal Metabolism in Cocaine Abusers: Implications in Addiction
Abstract
OBJECTIVE: The authors have shown that decreases in dopamine D2 receptors in cocaine abusers were associated with decreased metabolism in the cingulate and prefrontal and orbitofrontal cortices. To assess whether increasing dopamine would reverse these metabolic decrements, they measured the effects of methylphenidate, a drug that increases dopamine, on brain glucose metabolism in 20 cocaine abusers. METHOD: The subjects underwent two [18F]fluorodeoxyglucose positron emission tomography scans, one after two sequential placebo injections and one after two intravenous doses of methylphenidate. D2 receptors were measured with [11C]raclopride to evaluate their relation to methylphenidate-induced metabolic changes. RESULTS: Methylphenidate induced variable changes in brain metabolism: subjects with the higher D2 measures tended to increase metabolism, whereas those with the lower D2 measures tended to decrease metabolism. Methylphenidate’s effects were significant for increases in metabolism in the superior cingulate, right thalamus, and cerebellum. Methylphenidate-induced changes in the right orbitofrontal cortex and right striatum were associated with craving, and those in the prefrontal cortex were associated with mood. CONCLUSIONS: Although methylphenidate increased metabolism in the superior cingulate, it only increased metabolism in orbitofrontal or prefrontal cortices in the subjects in whom it enhanced craving and mood, respectively. This indicates that dopamine enhancement is not sufficient per se to increase metabolism in these frontal regions. Activation of the right orbitofrontal cortex and right striatum (brain regions found to be abnormal in compulsive disorders) in the subjects reporting craving may be one of the mechanisms underlying compulsive drug administration in addicted persons. The predominant correlation of craving with right but not left brain regions suggests laterality of reinforcing and/or conditioned responses.
The ability of cocaine to increase the extracellular concentration of dopamine in the nucleus accumbens, a brain region associated with drug reinforcement, has been postulated to be crucial for cocaine’s reinforcing effects (1, 2). However, cocaine’s actions on other brain regions receiving projections from the mesocortical, mesolimbic, or mesostriatal dopamine pathways are also likely to be involved in its reinforcing and addictive effects. Although it has been postulated that cocaine addiction results from a decrease in brain dopamine (3), the mechanism (or mechanisms) by which a dysfunctional dopamine system would lead to addiction is poorly understood. Using positron emission tomography (PET) and a multiple-tracer approach to evaluate the dopamine system in cocaine abusers, we have documented significant decrements in dopamine D2 receptors, which persisted after protracted detoxification and which were associated with decreased metabolic activity in the prefrontal cortex, cingulate gyrus, and orbitofrontal cortex (4). Because we had also shown that the orbitofrontal cortex and the striatum were hypermetabolic in active cocaine abusers reporting intense craving (5), we postulated that during cocaine intoxication, the increase in dopamine facilitates activation of these brain regions, which leads to the craving and subsequent impulsive and compulsive drug intake characteristic of the addicted individual (6).
To test this hypothesis, we measured the effects of two sequential doses of methylphenidate on regional brain glucose metabolism in 20 detoxified cocaine abusers. We used methylphenidate, a psychostimulant drug that increases dopamine by blocking the dopamine transporters (7), because it induces a sustained and long-lasting inhibition of dopamine transporters in the brain (half-life >90 minutes) (8). We measured regional brain glucose metabolism by means of PET and [18F]fluorodeoxyglucose (FDG). Measures of dopamine D2 receptors were also obtained in 19 of the subjects with the use of [11C]raclopride in order to evaluate the contribution that D2 baseline measures made to the metabolic responses of the subjects. Methylphenidate has pharmacological properties similar to those of cocaine (8), and when it is injected intravenously, cocaine abusers report that it has effects similar to those of intravenous cocaine (9). Two sequential doses of methylphenidate were given rather than a single dose, since drug abuse is characterized by repeated drug administration. We hypothesized that methylphenidate would increase metabolic activity in the prefrontal cortex, cingulate gyrus, and orbitofrontal cortex and that it would increase metabolism in the striatum and the orbitofrontal cortex in proportion to the magnitude of the craving.
METHOD
The subjects were 20 male right-handed inpatients at the VA Medical Center, Northport, N.Y. (mean age=36 years, SD=5; mean number of years of education=12, SD=2; mean verbal IQ=103, SD=11). The subjects met the DSM IV criteria for cocaine dependence and had used cocaine (freebase or crack), at least 4 grams a week, for at least the preceding 6 months. The exclusion criteria were current or past psychiatric disease other than cocaine dependence; past or present history of neurological, cardiovascular, or endocrinological disease; history of head trauma; current medical illness; and dependence on any substance other than cocaine, nicotine, or caffeine. Subjects were tested within 1 month of last cocaine use (mean=14 days, SD=7) and had an average history of 13 years (SD=5) of cocaine use. After complete description of the study to the subjects, written informed consent was obtained.
Each subject underwent two PET FDG scans done on separate days. The first was done 7 minutes after two sequential placebo injections (given 90 minutes apart), and the second was done 7 minutes after two sequential methylphenidate doses (0.50 and 0.25 mg/kg i.v. given 90 minutes apart). The order was fixed to avoid possible lasting effects of methylphenidate on brain metabolism. Subjects were blind to the drugs received. Nineteen of the subjects also underwent a [11C]raclopride scan, which was conducted 2 hours before the baseline metabolic scan. For the metabolic scan, emission scans were started 35 minutes after injection of 4–6 mCi of FDG and were obtained for 20 minutes on a CTI 931 tomograph (6.5 mm × 5.9 mm × 5.9 mm full width at half maximum, 15 slices) with the use of previously described procedures (10). For the D2 scans, sequential emission scans were obtained immediately after intravenous injection of 4–10 mCi of [11C]raclopride (specific activity=0.5–1.5 Ci/mM at end of bombardment) and continued for a total of 60 minutes as described previously (11).
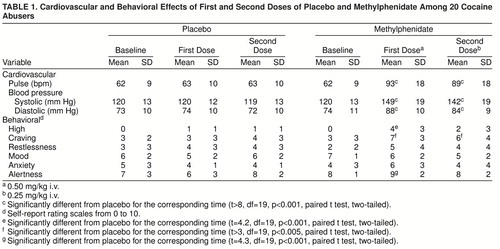
Prior to placebo and/or methylphenidate and 27 minutes after the first and the second methylphenidate doses, subjects recorded self-reports for drug effects—the “high,” craving (averaged scores on desire for cocaine and on perception of loss of control over cocaine), restlessness, mood (continuum between depressed and happy), anxiety, and alertness—using verbal analog scales from 0 (no effect) to 10 (maximal effect) (9). Heart rate and systolic and diastolic blood pressure were monitored continuously throughout the procedure. Methylphenidate concentration in plasma was measured by means of capillary gas chromatography/mass spectrometry.
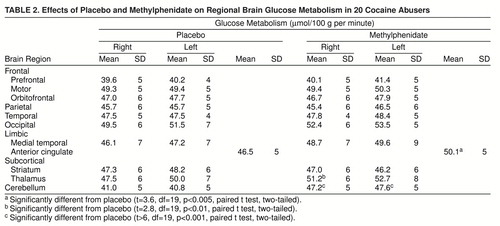
To select the regions in the metabolic images, we used a template, which we had previously described (10), that locates 114 regions of interest. We averaged the values of the regions of interest from the different slices corresponding to the same anatomic regions into 21 “composite” brain regions including one for the whole brain (the average of metabolism in 15 brain slices). We compressed the original template into 21 regions because this significantly increases the reproducibility of our measures and decreases the multiplicity of comparisons. Metabolic measures were also normalized in relation to the activity in the whole brain to generate “relative” measures. Relative measures were obtained because they may be more sensitive than “absolute” measures for detecting regional drug effects when the drug induces significant changes in global metabolism. Hypothesis-driven regions (the orbitofrontal cortex, prefrontal cortex, and cingulate gyrus) were selected on the basis of our previous results showing a correlation between metabolic activity in these brain regions and dopamine D2 receptors. In addition, for the exploratory analysis, we chose a representative sample of regions in the cortical lobes, subcortical structures, and cerebellum. For the [11C]raclopride images, the time-activity curves in the striatum and in the cerebellum and the time-activity curves for unchanged tracer in plasma were used to obtain distribution volumes (12). The ratio of the distribution volume in the striatum to that in the cerebellum, which corresponds to Bmax/Kd+1 and is insensitive to changes in cerebral blood flow (CBF), was the measure for D2 receptor availability (13).
Effects of methylphenidate on the absolute and the relative metabolic measures were tested with paired t tests. Pearson product-moment correlations between the regional changes in metabolism (from placebo to methylphenidate) and the D2 measures and methylphenidate-induced behavioral changes were performed. For the behavioral changes, we used the ratings from the second placebo dose and the ratings from the second methylphenidate dose (change from placebo 2 to methylphenidate 2), since those were the measurements taken at the time of the FDG scan. For hypothesis-driven correlations, significance was set at p<0.05, and for the others, significance was set at p<0.01.
Effects of methylphenidate on the behavioral and cardiovascular measures were tested by means of paired t tests in which the score obtained during placebo for the equivalent time measurement was used.
RESULTS
The mean serum concentration of methylphenidate was 107 ng/ml (SD=28) 27 minutes after administration of the first methylphenidate dose and 118 ng/ml (SD=31) 37 minutes after the second methylphenidate dose. Methylphenidate induced significant changes in the cardiovascular measures: both doses significantly increased pulse rate and systolic and diastolic blood pressure (table 1). Methylphenidate also induced significant changes in the behavioral measures: both doses significantly increased the craving for cocaine, whereas only the first dose increased alertness and the high (table 1).
Methylphenidate did not change global glucose metabolism (placebo mean=35.1 µmol/100 g per minute, SD=4; methylphenidate mean=36.2 µmol/100 g per minute, SD=3). This was because of the variability in responses: methylphenidate increased global metabolism more than 5% in eight subjects, decreased it more than 5% in seven subjects, and did not change it in five subjects. Analysis of the changes in the regional metabolic measures showed significant methylphenidate-induced increases in the right and left cerebellum, the superior cingulate gyrus, and the right thalamus (table 2). Correlation analysis of the measures of dopamine D2 receptor availability were significant for methylphenidate-induced changes in global metabolism (r=0.69, df=18, p<0.002). Subjects with the higher D2 measures tended to show global increases in metabolism with methylphenidate, whereas those with low D2 measures tended to show decreases (figure 1). Correlations between D2 measures and the changes in the regional measures were significant for most cortical brain regions and for the cerebellum (table 3).
Methylphenidate’s effects on the relative metabolic measures were significant for increases in the right and left cerebellum (t>8.7, df=19, p<0.001) and in the superior cingulate gyrus (t=3.6, df=19, p<0.002) and for decreases in the left striatum (t=3.4, df=19, p<0.004) (data not shown).
The hypothesis-driven correlations between methylphenidate-induced changes in craving and changes in the absolute metabolic measures were significant for the right striatum (r=0.61, df=19, p<0.005) and the right orbitofrontal cortex (r=0.52, df=19, p<0.02). A similar pattern was observed for the relative metabolic measures, with significant correlations between craving and the right striatum (r=0.73, df=19, p<0.001) and the right orbitofrontal cortex (r=0.64, df=19, p<0.003) (figure 2). Subjects who experienced intense craving had increased absolute and relative metabolism in the right striatum and right orbitofrontal cortex with methylphenidate (figure 3).
We also found significant correlations between methylphenidate-induced changes in mood and changes in absolute metabolic activity in the right prefrontal cortex (r=0.60, df=19, p<0.005) and the left prefrontal cortex (r=0.68, df=19, p<0.001); increases in mood were associated with increases in prefrontal metabolism. The correlations between mood and prefrontal metabolism were not significant for the relative metabolic measures. None of the correlations with self-reports of high, restlessness, anxiety, and alertness reached significance. There were no correlations between plasma levels of methylphenidate and methylphenidate-induced changes in metabolism.
DISCUSSION
We recently reported that in normal subjects, two sequential doses of the psychostimulant drug methylphenidate induced variable metabolic responses, with some subjects showing increases in metabolism, some showing decreases, and some showing no change (14). Furthermore, we showed that an individual’s metabolic responses were correlated with his measures of dopamine D2 receptors (14). In the current study we found a similar variability among cocaine abusers and a similar correlation with dopamine D2 receptors; subjects with the higher D2 measures tended to have increased metabolism, whereas those with the lower D2 measures tended to have decreased metabolism. It is interesting that results of a study evaluating the effects of an acute dose of cocaine—a psychostimulant drug that, like methylphenidate, binds to the dopamine transporter—differ from the results of the current study in that cocaine consistently produced a decrease in brain glucose metabolism (15). Factors accounting for the differences between the results of that study and the current one are not known but are likely to reflect, among others, pharmacological differences between cocaine and methylphenidate (8), differences in drug administration (one dose of cocaine versus two doses of methylphenidate), and characteristics of the subjects investigated.
Although the normal subjects and the cocaine abusers showed many similarities in response to methylphenidate, there were also differences. The cocaine abusers, in contrast to the normal subjects, showed significant methylphenidate-induced increases in the superior cingulate gyrus and in the right thalamus. The cingulate gyrus has been linked with motivation and goal-directed behaviors (16), and thus its activation by dopamine may be one of the mechanisms by which dopamine modulates drive (17). Activation of the superior cingulate gyrus in the cocaine abusers could reflect the enhanced salience of the effects of methylphenidate, which cocaine abusers report to be similar to those of cocaine (9), in these subjects but not in control subjects. In this respect it is interesting to note that activation of the cingulate gyrus in cocaine abusers has been reported after administration of cocaine (18) and during cue-induced cocaine craving (19). Activation of the cingulate gyrus has also been reported in marijuana abusers during marijuana intoxication (20). The significance of the activation of the right thalamus is less clear, since very little is known about the involvement of the thalamus in drug-addictive behaviors. It is worth mentioning that we recently documented an enhanced responsivity to methylphenidate-induced changes in [11C]raclopride binding in the thalamus of cocaine abusers when compared to control subjects, and this enhanced sensitivity was associated with drug craving (21). We interpreted this abnormal thalamic response to methylphenidate as reflecting disrupted dopamine regulation of the striatal-thalamo-orbitofrontal cortex circuit in cocaine abusers.
Although we had hypothesized that in addition to increasing metabolism in the cingulate, methylphenidate would increase metabolism in the prefrontal and orbitofrontal cortex, metabolic activation in these latter two brain regions was only observed in subjects for whom methylphenidate enhanced mood and in those for whom it induced intense craving, respectively. This indicates that elevation of dopamine is not sufficient per se to activate these frontal brain regions. Previous imaging studies had shown activation of the cingulate gyrus as well as the prefrontal cortex after administration of the dopamine agonist apomorphine (22, 23). Because the prefrontal cortex has been implicated in the regulation of mood (24), failure to observe a consistent activation of the prefrontal cortex by methylphenidate could reflect the variable effects of methylphenidate on mood, enhancing mood (shifting ratings toward happy) in some subjects while decreasing it (shifting ratings toward depressed) in others.
The orbitofrontal cortex processes information about aversive and reinforcing stimuli (25) and is involved in modifying an animal’s behavior when the reinforcing characteristics of these stimuli change (26). Thus, the differential activation of the orbitofrontal cortex in subjects who reported intense craving could reflect its participation as a function of the perceived reinforcing effects of methylphenidate. Activation of the right orbitofrontal cortex has also been reported during conditioned stimulation, a finding that was interpreted as reflecting the involvement of the orbitofrontal cortex in expectation of the stimulus (27). Thus, its activation in subjects in whom methylphenidate induced craving could reflect the subjects’ expectation of receiving another dose of methylphenidate, and this expectation may be consciously perceived as craving. That the correlation with craving was observed in the orbitofrontal cortex as well as in the striatum is likely to reflect the neuroanatomic connections between these two brain regions (28). In fact, behavioral deficits have been reported after lesioning of the orbitofrontal cortex that are similar to those seen after lesioning of the striatum (29). Lesions in either of these brain regions results in perseveration and resistance to extinction of reward-associated behaviors (30). In humans, pathology in the orbitofrontal cortex and in the striatum has been reported in patients with obsessive-compulsive disorders (31), which share with addiction the compulsive quality of the behavior. Thus, activation of the right orbitofrontal cortex and striatum by methylphenidate, a drug pharmacologically similar to cocaine, may be one of the mechanisms by which cocaine elicits craving and the subsequent compulsive drug administration in addicted individuals.
The correlation between craving and right but not left brain regions was unexpected. Although the literature is very limited, there is some evidence that there are left-right differences in drug responses; for example, craving for alcohol has been associated with increases in CBF in the right caudate (32), a predominant effect on the right has been reported in brain regions (cingulate, putamen, insula) where the activation induced by cocaine was temporally correlated with euphoria (18), and a predominant activation of right brain regions during marijuana intoxication has been reported (20). Also, as noted above, activation during conditioned stimulation was obtained for the right but not for the left orbitofrontal cortex (27). Further studies are required in order to evaluate whether there are left-right brain differences in response to reinforcing drugs and/or conditioned responses.
The metabolic correlates with craving that we observed differ from the findings that have been reported by other PET studies which used cocaine-cue videotapes designed to elicit craving. One of these studies (33) reported correlations between metabolic activation of the dorsolateral prefrontal cortex, medial temporal lobe (amygdala), and cerebellum and self-reports of craving, whereas another (34) reported that craving was associated with increases in relative CBF in the amygdala and anterior cingulate and with decreases in the basal ganglia. The reasons for the discrepancies between these three PET studies probably reflect the differences in PET methods (measuring glucose metabolism versus blood flow and measuring absolute versus relative activity), methods for eliciting craving, selection of brain regions (e.g., we did not measure the amygdala because of the errors in quantifying metabolism in such a small brain region with the limited spatial resolution of our PET scanner), and differences in the subjects investigated. A particular difference was that the current study gave a pharmacological challenge to increase dopamine and assess the effects of dopamine increases on frontal metabolism and craving, whereas the other two studies were designed to elicit craving by exposing the subjects to cocaine cues and observing the pattern of brain activation associated with the craving. Thus, while both strategies elicited craving, it is expected that the magnitude, duration, and association with other behavioral effects differed significantly for these methods.
The striatal D2 measures were not correlated with changes in striatal metabolism but mostly with changes in cortical and cerebellar metabolism. This probably reflects the fact that metabolic responses predominantly reflect activity in terminal regions (35), and thus the metabolic changes should be expected to occur in projection areas as well as in areas involved with secondary responses. This would explain the significant correlations between D2 measures and changes in the cerebellum, a brain region devoid of dopamine projections but neuroanatomically connected with the striatum (36). The cerebellum was also the brain region where methylphenidate induced the largest metabolic changes. Cerebellar activation has been reported to occur not only with other psychostimulants (37) but also during marijuana intoxication (38), and it has been associated with craving induced by cocaine cues (33). Although the cerebellum is mainly associated with motor effects, there is evidence that it may participate in the reinforcing properties of natural as well as pharmacological stimuli (39, 40). The cerebellum has connections with limbic brain regions (41), so that cerebellar activation could, through its neuroanatomic connections, activate regions directly involved with reward.
The failure to observe a correlation between regional brain changes induced by methylphenidate and self-reports of a high is most likely due to the fact that the high induced by the second methylphenidate dose was minimal, and the reports were obtained 27 minutes after methylphenidate administration, at which time the peak behavioral effects for the high had already occurred. The relatively poor temporal resolution of the FDG method (20–25 minutes) limits its sensitivity for detecting activation patterns associated with short-lasting drug effects such as the high. Even for longer-lasting effects of methylphenidate such as changes in craving and mood, for which correlations were observed, it is important to realize that the behaviors only reflected one temporal measure, and their intensity may have varied over the 20- to 25-minute period of FDG uptake. On the other hand, the FDG method has the advantage over methods that rely on blood flow changes (functional magnetic resonance imaging and [15O]water) in not having the confounding factor that is introduced by the vasoactive effects of many of the psychoactive drugs. The correlation between mood and changes in prefrontal metabolism was observed for the absolute metabolic measures but not for the relative measures, and we therefore cannot rule out the possibility that this correlation is significantly driven by global changes.
Variables confounding the interpretation of these results relate to the conditions of the study; subjects had expectations of receiving a psychostimulant drug for the baseline scan (placebo), which in itself may have activated prefrontal and orbitofrontal regions, making the effects of methylphenidate less apparent. Also, methylphenidate’s metabolic effects are likely to reflect not only its dopamine but also its noradrenergic effects (8). Finally, recovery of metabolism in frontal brain regions may require longer dopamine stimulation than that induced by two methylphenidate doses.
In summary, this study shows that dopamine enhancement is not sufficient per se to increase frontal metabolism. It also shows an association between drug craving and the right orbitofrontal cortex and right striatum. The striato-orbitofrontal circuit is involved with the salience of reinforcing stimuli, and thus its activation may be one of the mechanisms associated with the loss of control and the compulsive drug administration observed in cocaine-addicted subjects. In parallel, the activation by dopamine of the cingulate gyrus, a brain region involved with drive and motivation, could contribute to the desire for more cocaine during cocaine-induced dopamine stimulation. The predominant correlation of craving with right but not left brain regions merits further investigation to determine whether there is laterality of reinforcing and/or conditioned drug responses.
Received Jan. 29, 1998; revision received June 23, 1998; accepted July 2, 1998. From the Medical and Chemistry Departments, Brookhaven National Laboratory; the Department of Psychiatry; State University of New York at Stony Brook; and the Department of Psychiatry, New York University, New York. Address reprint requests to Dr. Volkow, Medical Department, Brookhaven National Laboratory, Bldg. 490, Upton, NY 11973; [email protected] (e-mail). Supported in part by U.S. Department of Energy contract DE-ACO2-76CH-00016 and National Institute on Drug Abuse grant DA-06891. The authors thank David Alexoff, Robert Carciello, Paula Cervany, Richard Ferrieri, Payton King, Alex Levy, Robert MacGregor, Noelwah Netusil, Carol Redvanly, David Schlyer, Colleen Shea, Donald Warner, and Cristopher Wong for their contributions.
 |
 |
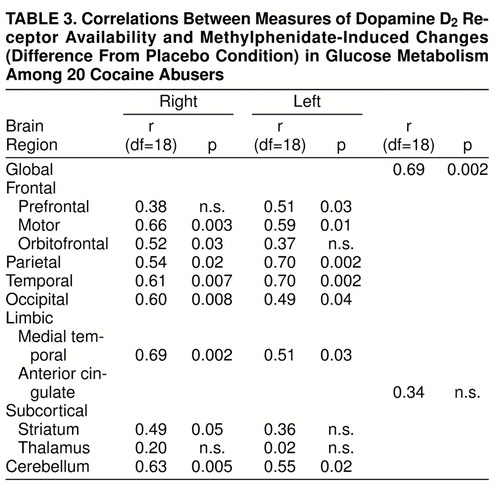 |

FIGURE 1. Regression Slopes Between Estimates of Dopamine D2 Receptor Availability (Bmax/Kd) Obtained With [11C]Raclopride and Metabolic Changes Induced by Methylphenidate (Difference From Placebo Condition) in the Whole Brain in a Study of Cocaine Abusersa
aPearson product-moment correlations, df=18.

FIGURE 2. Regression Slopes Between Methylphenidate-Induced Changes in Relative Metabolic Activity in the Right Striatum and the Right Orbitofrontal Cortex and Changes in Self-Reports of Craving in a Study of Cocaine Abusersa
aPearson product-moment correlations, df=19.

FIGURE 3. Brain Metabolic Images at the Level of the Striatum and the Orbitofrontal Cortex After Placebo and After Methylphenidate for a Cocaine Abuser Who Experienced High Levels of Craving and for One Who Experienced Low Levels of Craving After Methylphenidatea
aNotice that the cocaine abuser who experienced craving had an increase in metabolism in the right orbitofrontal cortex (R OFC) and in the right striatum, including the right caudate and right putamen (R PUT)
1. Koob G, Bloom FE: Cellular and molecular mechanisms of drug dependence. Science 1988; 242:715–723Crossref, Medline, Google Scholar
2. Di Chiara G, Imperato A: Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 1988; 85:5274–5278Crossref, Medline, Google Scholar
3. Dackis CA, Gold, MS: New concepts in cocaine addiction: the dopamine depletion hypothesis. Neurosci Biobehav Rev 1985; 9:469–477Crossref, Medline, Google Scholar
4. Volkow ND, Fowler JS, Wang G-J, Hitzemann R, Logan J, Schlyer D, Dewey S, Wolf AP: Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 1993; 14:169–177Crossref, Medline, Google Scholar
5. Volkow ND, Fowler JS, Wolf AP, Hitzemann R, Dewey S, Bendriem B, Alpert R, Hoff A: Changes in brain glucose metabolism in cocaine dependence and withdrawal. Am J Psychiatry 1991; 148:621–626Link, Google Scholar
6. Volkow ND, Ding Y-S, Fowler JS, Wang G-J: Cocaine addiction: hypothesis derived from imaging studies with PET. J Addict Dis 1996; 15:55–71Crossref, Medline, Google Scholar
7. Butcher SP, Liptrot J, Aburthnott GW: Characterisation of methylphenidate and nomifensine induced dopamine release in rat striatum using in vivo brain microdialysis. Neurosci Lett 1991; 122:245–248Crossref, Medline, Google Scholar
8. Volkow ND, Ding Y-S, Fowler JS, Wang G-J, Logan J, Gatley JS, Dewey SL, Ashby C, Lieberman J, Hitzemann R, Wolf AP: Is methylphenidate like cocaine? studies on their pharmacokinetics and distribution in human brain. Arch Gen Psychiatry 1995; 52:456–463Crossref, Medline, Google Scholar
9. Wang G-J, Volkow ND, Hitzemann R, Wong C, Angrist B, Burr G, Pascani K, Pappas N, Lu A, Cooper T, Lieberman J: Behavioral and cardiovascular effects of intravenous methylphenidate in normal subjects and cocaine abusers. Eur Addict Res 1997; 3:49–54Crossref, Google Scholar
10. Wang G-J, Volkow ND, Roque C, Cestaro V, Hitzemann R, Cantos E, Levy AV, Wolf AP: Functional significance of ventricular enlargement and cortical atrophy in normals and alcoholics as assessed by PET, MRI and neuropsychological testing. Radiology 1992; 186:59–65Crossref, Google Scholar
11. Volkow ND, Fowler JS, Wang G-J, Dewey SL, Schlyer D, MacGregor R, Logan J, Alexoff D, Shea C, Hitzemann R, Angrist B, Wolf AP: Reproducibility of repeated measures of 11C raclopride binding in the human brain. J Nucl Med 1993; 34:609–613Medline, Google Scholar
12. Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer D, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, Christman DR: Graphical analysis of reversible radioligand binding from time-activity measurements. J Cereb Blood Flow Metab 1990; 10:740–747Crossref, Medline, Google Scholar
13. Logan J, Volkow ND, Fowler JS, Wang G-J, Dewey SL, MacGregor R, Schlyer D, Gatley SJ, Pappas N, King P, Hitzemann R, Vitkun S: Effects of blood flow on [11C]raclopride binding in the brain: model simulations and kinetic analysis of PET data. J Cereb Blood Flow Metab 1994; 14:995–1010Crossref, Medline, Google Scholar
14. Volkow ND, Wang G-J, Fowler JS, Logan J, Angrist B, Hitzemann RJ, Lieberman J, Pappas NS: Effects of methylphenidate on regional brain glucose metabolism in humans: relationship to dopamine D2 receptors. Am J Psychiatry 1997; 154:50–55Link, Google Scholar
15. London ED, Cascella NG, Wong DF, Phillips RL, Dannals RF, Links JM, Herning R, Grayson R, Jaffe JH, Wagner HN Jr: Cocaine-induced reduction of glucose utilization in human brain: a study using positron emission tomography and [fluorine18]-fluorodeoxyglucose. Arch Gen Psychiatry 1990; 47:567–574Crossref, Medline, Google Scholar
16. Devinsky O, Morrell MJ, Vogt BA: Contributions of anterior cingulate cortex to behaviour. Brain 1995; 118:279–306Crossref, Medline, Google Scholar
17. Kiyatkin EA: Functional significance of mesolimbic dopamine. Neurosci Biobehav Rev 1995; 19:573–598Crossref, Medline, Google Scholar
18. Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, Goodman JM, Kantor HL, Gastfriend DR, Riorden JP, Mathew RT, Rosen BR, Hyman SE: Acute effects of cocaine on human brain activity and emotion. Neuron 1997; 19:591–611Crossref, Medline, Google Scholar
19. Maas LC, Lukas SE, Kaufman MJ, Weiss RD, Daniels SL, Rogers VW, Kukes TJ, Renshaw PF: Functional magnetic resonance imaging of human brain activation during cue-induced cocaine craving. Am J Psychiatry 1998; 155:124–126Link, Google Scholar
20. Mathew RJ, Wilson WH, Coleman E, Turkington TG, DeGrado TR: Marijuana intoxication and brain activation in marijuana smokers. Life Sci 1997; 60:2075–2089Crossref, Medline, Google Scholar
21. Volkow ND, Wang G-J, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Pappas N: Decreased striatal dopaminergic responsivity in detoxified cocaine abusers. Nature 1997; 386:830–833Crossref, Medline, Google Scholar
22. Grasby PM, Friston KJ, Bench CJ, Cowen PJ, Frith CD, Liddle PF, Frackowiak RS, Dolan RJ: The effect of the dopamine agonist, apomorphine, on regional cerebral blood flow in normal volunteers. Psychol Med 1993; 23:605–612Crossref, Medline, Google Scholar
23. Kapur S, Meyer J, Wilson AA, Houle S, Brown GM: Activation of specific cortical regions by apomorphine: a [15O]H2O PET study in humans. Neurosci Lett 1984; 176:21–24Crossref, Google Scholar
24. Kennedy SH, Javanmard M, Vaccarino FJ: A review of functional neuroimaging in mood disorders: positron emission tomography and depression. Can J Psychiatry 1997; 5:467–475Google Scholar
25. Aou S, Oomura Y, Nishino H, Inokuchi A, Mizuno Y: Influence of catecholamines on reward-related neuronal activity in monkey orbitofrontal cortex. Brain Res 1983; 267:165–170Crossref, Medline, Google Scholar
26. Thorpe SJ, Rolls ET, Maddison S: The orbitofrontal cortex: neuronal activity in the behaving monkey. Exp Brain Res 1983; 49:93–115Crossref, Medline, Google Scholar
27. Hugdahl K, Berardi A, Thompson WL, Kosslyn SM, Macy R, Baker DP, Alpert NM, LeDoux JE: Brain mechanisms in human classical conditioning: a PET blood flow study. Neuroreport 1995; 6:1723–1728Crossref, Medline, Google Scholar
28. Johnson T, Rosvold HE, Mishkin M: Projections from behaviorally defined sectors of the prefrontal cortex to the basal ganglia, septum and diencephalon of the monkey. J Exp Neurol 1968; 21:20–34Crossref, Google Scholar
29. Divac I, Rosvold HE, Szwarcbart MK: Behavioral effects of selective ablation of the caudate nucleus. J Comp Physiol Psychol 1967; 63:184–190Crossref, Medline, Google Scholar
30. Johnson TN: Topographic projections in the globus pallidus and the substantia nigra of selectively placed lesions in the precommissural caudate nucleus and putamen in the monkey. Exp Neurol 1971; 33:584–596Crossref, Medline, Google Scholar
31. Insel TR: Towards a neuroanatomy of obsessive-compulsive disorder. Arch Gen Psychiatry 1992; 49:739–744Crossref, Medline, Google Scholar
32. Modell JG, Mountz JM: Focal cerebral blood flow change during craving for alcohol measured by SPECT. J Neuropsychiatry Clin Neurosci 1995; 7:15–22Crossref, Medline, Google Scholar
33. Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C, Phillips RL, Kimes AS, Margolin A: Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci USA 1996; 93:12040–12045Crossref, Medline, Google Scholar
34. Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP: Limbic activation during cue-induced cocaine craving. Am J Psychiatry 1999; 156:11–18Link, Google Scholar
35. Schwartz WJ, Smith CB, Davidsen L, Savaki H, Sokoloff L, Mata M, Fink DJ, Gainer H: Metabolic mapping of functional activity in the hypothalamo-neurohypophysial system of the rat. Science 1979; 205:723–725Crossref, Medline, Google Scholar
36. Hook JC, Wise SP: Distributed modular architecture linking basal ganglia, cerebellum and cerebral cortex: their role in planning and controlling action. Cereb Cortex 1995; 2:95–110Crossref, Google Scholar
37. Ernst M, Zametkin AJ, Matochik J, Schmidt M, Jons PH, Liebenauer LL, Hardy KK, Cohen RM: Intravenous dextroamphetamine and brain glucose metabolism. Neuropsychopharmacology 1996; 17:391–401Crossref, Google Scholar
38. Volkow ND, Gillespie H, Mullani N, Tancredi L, Grant C, Valentine A, Hollister L: Brain glucose metabolism in chronic marijuana users during baseline and during marijuana intoxication. Psychiatry Res 1996; 67:29–38Crossref, Medline, Google Scholar
39. Plotnik R, Mir D, Delgado JMR: Map of reinforcing sites in the rhesus monkey brain. Int J Psychobiology 1972; 2:1–21Google Scholar
40. Ball GG, Micco DJ, Berntson GG: Cerebellar metabolism in the rat: complex stimulation-bound oral behavior and self-stimulation. Physiol Behav 1974; 13:123–127Crossref, Medline, Google Scholar
41. Heath RG, Harper JW: Ascending projection of the cerebellar fastigial nucleus to the hyppocampus, amygdala and other temporal lobe sites: evoked potential and histological studies in monkeys and cats. Exp Neurol 1974; 45:268–287Crossref, Medline, Google Scholar

