
Effect of a Haloperidol Challenge on Regional Brain Metabolism in Neuroleptic-Responsive and Nonresponsive Schizophrenic Patients
Abstract
OBJECTIVE: The CNS metabolic response to a neuroleptic challenge in treatment-responsive and nonresponsive schizophrenic patients was measured in order to examine the relation between treatment outcome and the capacity to alter neurochemical function in response to acute receptor blockade. METHOD: Positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG) were used to measure regional cerebral metabolism in seven schizophrenic patients judged to have been responsive to drug treatment previously and seven nonresponsive schizophrenic patients after a drug-free period of at least 3 weeks (baseline) and again 12 hours after administration of 5.0 mg of haloperidol. RESULTS: The haloperidol challenge caused widespread decreases in absolute metabolism in the nonresponsive patients but not the responsive patients. These group differences reflect the findings on the second (challenge) scans, since metabolic values at baseline were not statistically different in the two groups. The pattern of decreased metabolic activity in the nonresponders after the haloperidol challenge is similar to that previously observed in normal subjects. CONCLUSIONS: The metabolic response to drug challenge separates treatment responders from nonresponders and normal subjects. The results suggest that subtyping of schizophrenia (and other psychiatric disorders) can be achieved by measuring the physiologic response to a pharmacologic challenge in vivo with chemical brain-imaging techniques.
The therapeutic effectiveness of typical antipsychotic medications is strongly correlated with their capacity to block dopamine D2 receptors (1). However, clinical improvement may not be evident until several days or weeks after the beginning of treatment (2), despite the fact that peak blockade of nigro~striatal D2 receptors occurs within a few hours after a single dose (3).
In the course of treatment with typical antipsychotic drugs, both treatment-responsive and nonresponsive schizophrenic patients have been shown to produce equivalent D2 receptor blockade (4, 5). From these studies, we conclude that failure to respond clinically to treatment is not a failure of drug delivery but a failure of drug activity. Little is known about neurochemical changes occurring after initial receptor blockade that result in clinical efficacy. However, it seems likely that patients who differ in their clinical response to neuroleptic drugs may also differ in their CNS metabolic response to this initial blockade.
To investigate this hypothesis we developed an assay to measure the response of the CNS to acute receptor blockade (6, 7), using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG) to measure the effect on cerebral glucose metabolism of a single dose of haloperidol (5.0 mg i.m.). This single dose has been shown to be sufficient to produce essentially complete dopamine receptor blockade (3). In the present study we compared the metabolic response to neuroleptic challenge in treatment-responsive and nonresponsive schizophrenic patients to examine the relation between treatment outcome and the capacity to alter neurochemical function in response to acute dopaminergic receptor blockade.
Previously, we examined the response to a 5.0-mg i.m. haloperidol challenge in young normal male volunteers scanned 2 hours or 12 hours after haloperidol administration. After 2 hours, few of the changes in regional glucose administration reached statistical significance (7). Twelve hours after haloperidol administration, there was significantly reduced glucose utilization in the neocortex, limbic cortex, thalamus, and caudate nucleus but not in the putamen or cerebellum (6). These studies show that the PET/FDG method provides a measure of functional response that is sensitive to the effects of a neuroleptic challenge in human subjects, since we have shown previously that the test-retest variation is less than 5% (6). These studies also demonstrate that the effects of a single blocking dose of haloperidol are evident in areas that are poor in D2 receptors (e.g., the frontal cortex) but also in the striatum, suggesting that the functional response to neuroleptic challenge involves an interaction of striatal dopaminergic neurons with other, nonstriatal and possibly nondopaminergic, systems. In the present study, the PET/FDG assay was used to measure metabolic response to a neuroleptic challenge in treatment-responsive and nonresponsive schizophrenic subjects.
METHOD
The study followed guidelines approved by the human studies institutional review boards and radiation safety boards of the New York University Medical Center, the Manhattan Psychiatric Center, and Brookhaven National Laboratory. After a complete description of the study to the subjects, written informed consent was obtained.
Fourteen patients (13 male and one female) whose sole active DSM-III-R axis I disorder was chronic schizophrenia were included in the study. All subjects were in good physical health according to history, physical examination, blood and urine chemistry, and magnetic resonance imaging (MRI) examination of the brain. At the time of the study, 12 subjects were inpatients. All were right-handed. Three subjects had never received neuroleptic medication. Characteristics of the study group are presented in table 1.
Classification into responder and nonresponder groups involved a specific examination of past history and current mental status. The latter was assessed after a medication-free period (“unmedicated state”) and again after a minimum of 4 weeks of treatment with a typical neuroleptic (“medicated state”). The 7-point, 18-item Brief Psychiatric Rating Scale (BPRS) (8) was administered before the beginning of the unmedicated state, every 7 days thereafter, and on the days of the baseline and challenge PET scans. Of the 14 patients in the study, eight were being treated with typical neuroleptic drugs at the time of their enrollment. With treatment, some had already experienced symptom improvement.
To qualify as a responder, a subject first had to have a medicated-state BPRS score less than 42 and clear evidence of substantially more severe symptoms while in the unmedicated state. For the patients being treated at the time of enrollment, a 20% increment from the medicated-state BPRS score after washout was accepted as evidence of substantially more severe symptoms. To be considered a nonresponder, a subject had to have either a medicated-state BPRS score greater than 50 or a medicated-state BPRS score greater than 42 and no clear evidence of more severe symptoms in the unmedicated state. For the subjects whose hospital records did not give a clear indication of treatment efficacy (three responders and three nonresponders), a prospective 4-week neuroleptic trial was initiated in order to provide a measure of psychopathology in both medicated and unmedicated states. This algorithm captured the salient distinction that some patients (nonresponders) remained clinically very ill and unchanged with treatment.
All subjects but one who were on a medication regimen at the time of recruitment had their medication withdrawn for a minimum of 21 days (mean=32 days) before the unmedicated-state baseline scan. The exception, who had been medication free for many weeks, received a single dose of neuroleptic just before recruitment and was thereafter kept medication free for a further 13 days. No medication was administered during the 72 hours immediately preceding PET scanning. Only three patients required treatment for agitation during the neuroleptic-free period (two received 1 mg of lorazepam; one received 500 mg of chloral hydrate).
During washout, some of the patients evidenced clinical decline, while others did not. The apparent complexity of the assignment of patients into treatment responder or nonresponder groups was due to the practical consideration that many potential subjects were treated and partially recompensated before we could enroll them in the study. In order to minimize the confounding effect of treatment, each potential subject was evaluated for potential treatment response and recent exposure to psychotropic medication. The subjects and their records were rated by at least two senior attending psychiatrists (one of whom was always P.S., a highly trained rater and research psychiatrist).
Following the medication-free period, all subjects underwent a baseline PET/FDG scan and a second scan administered 12 hours after they received a 5.0-mg i.m. dose of haloperidol (mean dose=0.07 mg/kg, SD=0.01). As in our study of normal subjects (6), no anticholinergics were required for alleviation of extrapyramidal side effects before the PET scans. In all subjects, plasma neuroleptic levels were undetectable at the time of the unmedicated baseline scan. The interval between baseline and challenge scans ranged from 1 to 7 days (mean=2 days). Blood samples for ascertaining plasma prolactin levels were obtained at the time of the baseline and challenge scans and analyzed with a radioimmunoassay kit (GammaCoat, Clinical Assays, Inc., Stillwater, Minn.). Subjects fasted overnight and were given a standard low-monoamine breakfast before each PET scan.
All PET/FDG scans were done at the Chemistry Department, Brookhaven National Laboratory, on a CTI 931 tomograph (15 slices; 128×128 matrix size; spatial resolution of 6×6×6.5 mm full width at half maximum) (6, 7). For each subject, baseline and challenge scans were done at approximately the same time of day. Subjects were scanned with their eyes open and ears unoccluded in a quiet, dimly lit room. Adequacy of counting statistics and plasma input function were ascertained for all scans. All data are expressed as micromoles of glucose per 100 grams of tissue per minute. Transmission scans were also done to ensure precise interscan alignment. All images were reformatted to the scan geometry of the first (baseline) PET scan. MRIs (obtained for each subject) were coregistered to the first (baseline) transmission scan by a surface-matching technique (6, 9). A principal axis method was used to coregister first and second PET scans (10).
Nonoverlapping regions of interest were defined on each subject's MRI by placing a 5×5-pixel box (39 mm2) in the approximate center of a chosen neuroanatomic structure on all slices in which the structure appeared (6). While the initial placement was defined by template, final placement was determined by a single experienced operator (E.J.B.) and transferred by computer software to the coregistered PET images. Regions of interest from individual slices were combined, as detailed in our previous study (6), into 16 large regions of interest. These encompassed the neocortex, which included the regions of interest from the left and right dorsolateral prefrontal cortex, temporal cortex, and occipital cortex. The mesial temporal cortex (which included the entorhinal cortex and hippocampus) encompassed another set of regions of interest, and the remaining regions of interest were from the striatum, thalamus, cerebellum, and anterior cingulate. A global (whole brain) glucose utilization rate was calculated by averaging the metabolic rates from all pixels within the skull above an 18-µmol threshold (our laboratory's average white matter value for schizophrenic patients minus one standard deviation).
The effect of this haloperidol challenge on regional glucose metabolism in drug-responsive and nonresponsive patients was examined in a series of analyses of variance (ANOVAs) with group (drug-responsive or drug-nonresponsive) as the between-subjects factor and condition (baseline or challenge) as the within-subject repeated measure. All statistical tests were performed with SYSTAT (11) and were two-tailed. A significance level of 0.05 (uncorrected for repeated measures) was adopted to increase sensitivity for the detection of differences between groups.
RESULTS
By definition, the drug-responsive and drug-nonresponsive patients differed in the severity of their symptoms after treatment. This difference was quite marked according to the total BPRS scores (table 1). Considering only BPRS items that measure positive symptoms (conceptual disorganization, hallucinations, suspiciousness, unusual thought content), the responsive patients had a mean medicated-state score of 9.4, whereas the nonresponsive patients had a mean medicated-state score of 16.4 (table 1). Thus, the two groups differed strikingly in their clinical response to neuroleptic treatment, especially along the positive symptom dimension.
An analysis of plasma prolactin values, available for five drug-responsive and five nonresponsive subjects, showed a significant main effect of condition (F=15.39, df=1,8, p=0.004) and no significant interactions. For both groups, prolactin values were significantly higher in response to the haloperidol challenge, indicating that for both groups, the drug resulted in functionally effective D2 receptor blockade, at least in the tuberoinfundibular system, and indicating that consistent with earlier findings (3–5), a parenteral challenge with 5.0 mg of haloperidol gives a high brain level of drug.
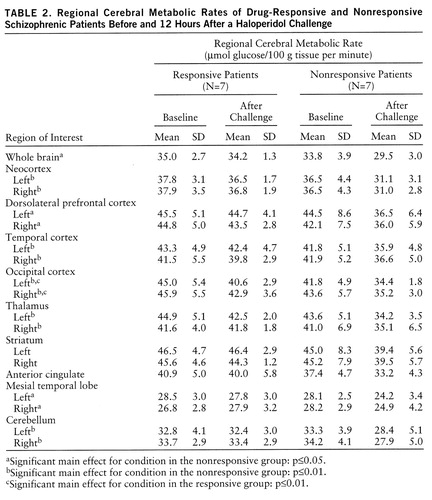
The single most striking effect of the haloperidol challenge (table 2) on whole brain metabolism was the significant decrease in glucose utilization in the drug-nonresponsive patients (F=8.78, df=1,6, p=0.03) but not in the drug-responsive patients (F=1.04, df=1,6, p=0.35) (figure 1). In the analysis by region of interest, there were significant main effects for group (responder, nonresponder) and condition (baseline, challenge) and a significant group-by-condition interaction. As can be seen in figure 1, the responders and nonresponders fell into clearly different groups, with the nonresponders showing decrements in every region assessed and the responders showing a significant decrease in the occipital lobe only. For comparison, the values of 12 normal subjects given the same challenge (6) and test-retest normal subjects (no challenge, N=9) are also displayed.
The neocortex was examined in its entirety and also divided into the dorsolateral prefrontal cortex and temporal and occipital cortexes. Analysis of simple effects (table 2) showed that neocortical metabolism was significantly reduced after challenge in the nonresponder group (F=12.53, df=1,6, p=0.01). Significant effects were also found for group (F=5.80, df=1,12, p=0.03) and condition (F=13.66, df=1,12, p=0.003), and there was a significant group-by-condition interaction (F=5.67, df=1,12, p=0.04). Regionally significant main effects and interactions in the neocortex were as follows: for the dorsolateral prefrontal cortex, condition (F=9.42, df=1,12, p=0.01) and group-by-condition interaction (F=5.27, df=1,12, p=0.04); for the temporal cortex, condition (F=13.25, df=1,12, p=0.003) and group-by-condition interaction (F=5.23, df=1,12, p=0.04); for the occipital lobe, only the main effect of condition (F=6.39, df=1,12, p=0.03). Post hoc ANOVA revealed no significant differences between the groups in whole brain metabolic rates at baseline (F=0.44, df=1,12, p=0.52).
Metabolic patterns of the patients categorized as responsive or nonresponsive on the basis of clinical history could not be distinguished from the results of the subjects (N=6) similarly classified on the basis of a prospective 4-week haloperidol trial. An ANOVA comparison of metabolic values for patients categorized by the two methods yielded no significant differences. For example, an ANOVA comparing neocortical values of prospectively (N=3) and retrospectively (N=4) categorized nonresponsive patients showed no significant main effect of categorization method (F=0.006, df=1,5, p=0.94) and no significant method-by-condition interaction (F=0.03, df=1,5, p=0.87). A similar ANOVA comparing neocortical values of prospectively (N=3) and retrospectively (N=4) categorized drug-responsive patients also showed no significant main effects. Limbic structures revealed a smaller effect; the anterior cingulate produced a not quite significant main effect of group (F=4.52, df=1,12, p=0.06) and a significant main effect of condition (F=5.37, df=1,12, p=0.04). The mesial temporal lobe did show a significant effect of condition (F=4.92, df=1,12, p=0.05) and a significant group-by-condition interaction (F=5.84, df=1,12, p=0.03).
There were no significant main effects or interactions in the stri~atum (table 2), indicating that glucose utilization in the two groups did not differ. The thalamus showed a significant effect of condition (F=14.36, df=1,12, p=0.003) and a significant group-by-condition interaction (F=8.05, df=1,12, p=0.02).
Analyses of cerebellar absolute metabolic response (table 2) showed a significant main effect of condition (F=13.39, df=1,12, p=0.003) and a significant group-by-condition interaction (F=10.40, df=1,12, p=0.007). A separate analysis of the neuroleptic-naive patients indicated a trend conforming to our overall results: two of the neuroleptic-naive patients responded to treatment (30-point and 17-point decreases in BPRS score) and had very little metabolic change in response to the challenge. The third neuroleptic-naive patient did not respond to treatment (5-point decrease) and had a significant decrease in metabolic activity in response to the challenge.
DISCUSSION
On the basis of our previous results demonstrating that binding of haloperidol was a necessary but not sufficient prerequisite for response to treatment in schizophrenic patients, we used PET and the FDG method to determine whether the regional cerebral metabolic response to a single haloperidol challenge could identify, upon admission to the hospital, the patients who would likely not respond to conventional neuroleptics. We expected that those who did not respond would fail to show a metabolic response consequent to a haloperidol challenge and that this would distinguish these patients from normal subjects and those who responded to treatment. We reasoned that those who responded would have a greater capacity than those who did not to alter neurochemical function in response to acute receptor blockade (haloperidol challenge), consistent with their characteristic decline in schizophrenic symptoms. Using this as evidence of a relatively active dopamine system, we hypothesized that responders to treatment would have metabolic rates similar to those of normal comparison subjects, who in our previous study demonstrated decreased metabolism in response to acute receptor blockade by haloperidol.
We found a similar prolactin response to the challenge in the two groups of schizophrenic patients, although the challenge caused widespread metabolic changes in nonresponsive (but not responsive) patients. The differences between groups were evidently a product of the challenge; baseline scans were virtually identical. While it was not in accord with our hypothesis, our finding of widespread decreased absolute metabolism in the nonresponsive patients is similar to our previous finding of significantly decreased absolute metabolism in young, normal male volunteers (6).
Following drug challenge, both normal subjects and nonresponsive schizophrenic patients evidenced significantly decreased absolute metabolism in most brain regions, although the changes in the striatum did not reach significance. Cerebellar changes were not significant in the normal group, although the very large decrement in cerebellar metabolism in the nonresponders is consistent with our prior report of decreased cerebellar metabolism in medicated chronic schizophrenic patients, many of whom were not responsive to treatment (12). This observation was noted previously under similar conditions (13) and deserves further study. Drug-responsive patients also showed significantly decreased absolute metabolism in the occipital region. This occipital effect has been observed in normal subjects and both drug-responsive and nonresponsive schizophrenic groups and most likely reflects diminished visual stimulation after challenge (14); i.e., many subjects found haloperidol to be sedating and had difficulty keeping their eyes open despite frequent reminders from the experimenter (6, 7).
Changes in plasma prolactin levels after the haloperidol challenge provide another measure of the responsiveness of the CNS to dopamine blockade. Prolactin levels are also an independent measure of the adequacy of the haloperidol challenge. Dopamine acts as an inhibitor of the synthesis and release of prolactin through D2 receptors in the tuberoinfundibular system (15). The increased plasma prolactin levels in both groups of patients after challenge indicate that haloperidol blocked D2 receptors sufficiently to disinhibit the synthesis and release of prolactin. The finding that the groups showed similar increases in prolactin might support the argument that prolactin release is not an indication of treatment efficacy.
The widespread metabolic effects observed in studies of normal and schizophrenic subjects may reflect effects occurring after D2 receptor blockade through projections from striatal and other D2-rich areas. It has been proposed that blockade of D2 receptors could propagate effects through cortical-striatal-thalamic pathways (16). Group differences in limbic, thalamic, and striatal response to a haloperidol challenge may reflect differences in these cortical-striatal-thalamic pathways and offer support for speculation that these pathways are involved in the pathophysiology of schizophrenia (17, 18).
Diminished metabolism in the group of nonresponders as compared with the responders certainly emphasizes that a lower metabolic rate does not necessarily imply lack of metabolic response or even a diminished capacity. As our data show, lower metabolic rates do not necessarily predict lack of metabolic response to a challenge and are not necessarily evidence of pathology (19). Our unmedicated schizophrenic patients could not be subcategorized at baseline solely on the basis of their metabolic pattern at rest. This is consistent with other studies that have shown that D2 receptor density does not distinguish normal control subjects from unmedicated schizophrenic patients (20).
Recent tracer kinetic studies by Laruelle et al. (21) and Breier et al. (22) have confirmed that in comparison with control subjects, schizophrenic subjects do not show elevated synaptic dopamine concentrations in vivo but do show elevated presynaptic dopamine reserves that can be mobilized with amphetamine. In the present study the ability of the haloperidol challenge to distinguish treatment-responsive schizophrenic patients from normal subjects and nonresponders to treatment is consistent with these findings if one considers a part of the metabolic response as reflecting the transitory mobilization of physiologic reserves of synaptic dopamine precursors.
What, then, is different about responders that enables them to tolerate a haloperidol challenge with neither metabolic decrements nor the appearance of untoward behaviors? One might consider the possibility that in healthy normal subjects and nonresponders, there is an inability to accommodate the temporary perturbation of tonic circuitry effected by haloperidol, while in responders the tonic pathophysiology differs in such a way that this perturbation does not cause a profound metabolic change. It is even possible that the cognitive and affective disturbances observed in normal subjects after a haloperidol challenge and observed in nonresponders despite exposure to this agent are consistent with a disturbance in autoregulation, since metabolic patterns do not distinguish responders from nonresponders either at baseline or after 4 weeks of treatment (23, 24).
These observations are consistent with a growing conception of the neurochemical basis of severe psychopathology that emphasizes maladaptation to perturbations rather than absolute levels of activity within the system (25). Of particular relevance is a model elaborated by Grace (26) that emphasizes the tonic modulation of phasic processes as an integral component of homeostatic mechanisms; this is discussed at length by Bilder (27) in a neural systems context. Decreased frontal metabolism could be indicative of the effects of haloperidol on glutamatergic afferents, thought to be involved in the release of low levels of presynaptic (tonic) dopamine into the extracellular fluid. Treatment responders might thus be able to accommodate the antidopaminergic effects of haloperidol in their phasic responses but nonresponders might not, presumably because of insufficient presynaptic physiologic reserve. This model postulates dysfunction of modulation in the manifestation of schizophrenic symptoms. Thus, if plasticity is central to all behavior and is restricted in pathological behavior (23), it is not necessary for an entire system to be affected, but merely for an integral component within that system, for whatever reason, to become maladaptive (27).
Although an acute challenge appears to distinguish treatment outcomes, several studies report that patterns of metabolic response to long-term treatment are similar in drug-responsive and nonresponsive patients. For example, Wolkin et al. (24) found no relation between change in clinical measures and change in regional metabolism after chronic haloperidol treatment. Holcomb et al. (28) obtained similar results, although it should be noted that their study group showed “surprisingly little” change in BPRS psychosis scores across the treatment interval and thus may have included few drug responders. Wolkin et al. (4) demonstrated that responsive and nonresponsive patients show similarly high levels of D2 receptor blockade after 4 weeks of treatment; Coppens et al. (5) showed similar results. In contrast, Buchsbaum and colleagues found that lower pretreatment striatal metabolism predicted a better clinical response to clozapine (29) and haloperidol (30) and that drug responders showed a greater increase in striatal metabolism after haloperidol therapy. However, these observations were not confirmed in related studies (23, 24). Nor did we find in the present study that baseline striatal metabolism differentiated our responder and nonresponder subjects (table 2).
These differences may reflect different study methods (e.g., our patients were scanned under resting conditions, whereas patients in the studies by Buchsbaum et al. [29, 30] were scanned while performing a visual monitoring task), small sample size, or differences in the spatial resolution of the various instruments. Overall, though, these studies suggest that drug-responsive and nonresponsive patients show a similar pattern of metabolic response to long-term drug administration.
Our classification algorithm relies primarily on our assessment of our subjects' symptoms when they are fully medicated. This classification is consistent with the experience of most clinicians. The usual clinical situation that prompts treatment with an atypical agent is residual symptom severity despite an adequate initial drug trial (nonresponse). Since chronic patients enter the hospital in various stages of decompensation and can show variable improvement, comparisons of decompensated state and medicated state are often inconsistent and unreliable because they depend on the severity of a given episode. Even a fixed period of washout does not ensure that a “reproducible” unmedicated state will ensue, since patients can maintain a meta~stable symptomatic state for varying periods. To delineate the groups more clearly, we excluded patients who remained substantially but not severely symptomatic after treatment (BPRS score >42 but <50), even though they had markedly more severe symptoms when they were not taking medication. Patients with medicated-state BPRS scores greater than 50 were judged to be sufficiently ill to be considered nonresponders irrespective of their unmedicated state. Our algorithm yielded groups that differed markedly in their clinical presentation after completion of the treatment protocol. Since errors in the clinical assignment would be expected to diminish rather than enhance the main effect, it is unlikely that our findings can be explained on this basis.
The purpose of this study was to determine whether the metabolic effects of acute receptor blockade differed in drug-responsive and nonresponsive patients to an extent that would allow prediction of responses. Even though the number of subjects was small, our findings were sufficiently robust to identify clear group differences, supporting the assumption that individual differences in the brain's neurochemical adaptation to acute receptor blockade may predict response to treatment in schizophrenia. This particular FDG haloperidol challenge paradigm may ultimately be shown to lack sufficient sensitivity to be immediately applicable in a clinical setting. Similar paradigms using specific neurotransmitter assays (23) may yet provide better discrimination than can be determined with FDG. In any case, the results of this study indicate that by measuring physiologic differences in the response to a pharmacologic challenge to the brain, there is not only the possibility of predicting appropriate treatment but, perhaps more important, of developing a psychiatric nosology based on physiology as well as behavior.
 |
 |
Received March 10, 1997; revision received Oct. 7, 1997; accepted Oct. 13, 1997. From the Department of Psychiatry and the Department of Radiology, New York University Medical Center, and the Department of Chemistry, Brookhaven National Laboratory, Upton, N.Y. Address reprint requests to Dr. Brodie, Department of Psychiatry, New York University Medical Center, 550 First Ave., New York, NY 10016. Supported by NMIH grant MH-47277; grant NS-15638 from the National Institute of Neurological Disorders and Stroke; grant RR-00096 from the National Center for Research Resources, NIH; a Senior Investigator Award from the National Alliance for Research on Schizophrenia and Depression to Dr. Brodie; contract DE-AC02-76CH00016 from the U.S. Department of Energy, Office of Health and Environmental Research, to Brookhaven National Laboratory; and a research fellowship grant from the Deutsche Forschungsgemeinschaft to Dr. Schl<148>sser. The authors thank Arlene Hurley and the nursing staff of the New York University-General Clinical Research Center and the Manhattan Psychiatric Center; E. Laska, M. Meissner, T. Cooper, W. Tsui, B. Tewelde, D. Schlyer, J. Fowler, C. Redvanly, C. Shea, D. Alexoff, P. King, D. Warner, N. Pappas, N. Netusil, T. Johnson, and H. Ulyat; and Drs. R. Bilder and A. Wagman for discussions. Dr. Bartlett died during the preparation of this manuscript. Her contribution to the understanding of schizophrenia did not.

FIGURE 1. Effect of a Haloperidol Challenge on Regional Glucose Metabolism in Drug-Responsive and Drug-Resistant Schizophrenic Patients and Normal Subjects
aSignificant interaction between group (responder, nonresponder) and condition (baseline, challenge): p<0.05.
1. Seeman P: Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987; 1:133–152Crossref, Medline, Google Scholar
2. Miller R: The time course of neuroleptic therapy for psychosis: role of learning processes and implications for concepts of psychotic illness. Psychopharmacology (Berl) 1987; 92:405–415Crossref, Medline, Google Scholar
3. Nordstrom A-L, Farde L, Halldin C: Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology (Berl) 1992; 106:433–438Crossref, Medline, Google Scholar
4. Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue C-Y, Cooper TB, Brodie JD: Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry 1989; 146:905–908Link, Google Scholar
5. Coppens H, Slooff C, Paans A, Wiegman T, Vaalburg W, Korf J: High central D2-dopamine receptor occupancy as assessed with positron emission tomography in medicated but therapy resistant schizophrenic patients. Biol Psychiatry 1991; 29:629–634Crossref, Medline, Google Scholar
6. Bartlett EJ, Brodie JD, Simkowitz P, Dewey SL, Rusinek H, Wolf AP, Fowler JS, Volkow ND, Smith G, Wolkin A, Cancro R: Effects of haloperidol challenge on regional cerebral glucose utilization in normal human subjects. Am J Psychiatry 1994; 151:681–686Link, Google Scholar
7. Bartlett EJ, Brodie JD, Simkowitz P, Dewey SL, Rusinek H, Vol~kow ND, Wolf AP, Smith G, Wolkin A, Cancro R: Time-dependent effects of a haloperidol challenge on energy metabolism in the normal human brain. Psychiatry Res 1996; 60:91–99Crossref, Medline, Google Scholar
8. Overall JE, Gorham DR: Introduction: the Brief Psychiatric Rating Scale: recent developments in ascertainment and scaling. Psychopharmacol Bull 1988; 24:97–99Google Scholar
9. Pelizzari CA, Chen GTY, Spelbring DR, Weichselbaum RR, Chen C: Accurate three-dimensional registration of CT, PET and/or MR images of the brain. J Comput Assist Tomogr 1989; 13:20–26Crossref, Medline, Google Scholar
10. Rusinek H, Tsui W-H, Levy AV, Noz ME, deLeon M: Principal axes and surface fitting methods for three-dimensional image registration. J Nucl Med 1993; 34:2019–2024Google Scholar
11. Wilkinson L: SYSTAT: The System for Statistics. Evanston, Ill, Systat, 1990Google Scholar
12. Volkow ND, Levy A, Brodie JD, Wolf AP, Cancro R, Van Gelder P, Henn F: Low cerebellar metabolism in medicated patients with chronic schizophrenia. Am J Psychiatry 1992; 149:686–688Link, Google Scholar
13. Andreasen NC, O'Leary DS, Cizadlo T, Arndt S, Rezai K, Boles Ponto LL, Watkins GL, Hichwa RD: Schizophrenia and cognitive dysmetria: a positron-emission tomography study of dysfunctional prefrontal-thalamic-cerebellar circuitry. Proc Natl Acad Sci USA 1996; 93:9985–9990Google Scholar
14. Phelps ME, Mazziotta JC, Kuhl DE, Nuwer M, Packwood J, Metter J, Engel J Jr: Tomographic mapping of human cerebral metabolism visual stimulation and deprivation. Neurology 1981; 31:517–529Crossref, Medline, Google Scholar
15. Moore KE: Hypothalamic dopaminergic neuronal systems, in Psychopharmacology: The Third Generation of Progress. Edited by Meltzer HY. New York, Raven Press, 1987, pp 127–139Google Scholar
16. Alexander GE, Crutcher MD, DeLong MR: Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res 1990; 85:119–146Crossref, Medline, Google Scholar
17. Siegel BV Jr, Buchsbaum MS, Bunney WE Jr, Gottschalk LA, Haier RJ, Lohr JB, Lottenberg S, Najafi A, Nuechterlein KH, Potkin SG, Wu JC: Cortical-striatal-thalamic circuits and brain glucose metabolic activity in 70 unmedicated male schizophrenic patients. Am J Psychiatry 1993; 150:1325–1336Google Scholar
18. Swerdlow NR, Koob GF: Dopamine, schizophrenia, mania and depression: toward a unified hypothesis of cortico-striato-pallido-thalamic function. Behavioral and Brain Sciences 1987; 10:197–245Crossref, Google Scholar
19. Brodie JD: Imaging for the clinical psychiatrist: facts, fantasies, and other musings. Am J Psychiatry 1996; 153:145–149Link, Google Scholar
20. Farde L, Wiesel FA, Hall H, Halldin C, Stone-Elander S, Sedvall G: No D2 receptor increase in PET study of schizophrenia. Arch Gen Psychiatry 1987; 44:671–672Crossref, Medline, Google Scholar
21. Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Soghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB: Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA 1996; 93:9235–9240Google Scholar
22. Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D: Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 1997; 94:2569–2574Google Scholar
23. Schloesser R, Simkowitz P, Bartlett EJ, Wolkin A, Smith GS, Dewey SL, Brodie JD: The study of neurotransmitter interactions using positron emission tomography and functional coupling. Clin Neuropharmacol 1996; 19:371–389Crossref, Medline, Google Scholar
24. Wolkin A, Sanfilipo M, Duncan E, Angrist B, Wolf AP, Cooper TB, Brodie JD, Laska E, Rotrosen JP: Blunted change in cerebral glucose utilization after haloperidol treatment in schizophrenic patients with prominent negative symptoms. Am J Psychiatry 1996; 153:346–354Link, Google Scholar
25. Friedhoff AJ, Simkowitz P: A new conception of the relationship between psychological coping mechanisms and biological stress buffering systems. Br J Psychiatry Suppl 1989; 4:61–66Medline, Google Scholar
26. Grace AA: Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 1991; 41:1–24Crossref, Medline, Google Scholar
27. Bilder RM: Neurocognitive impairment in schizophrenia and how it affects treatment options. Can J Psychiatry 1997; 42:255–264Crossref, Medline, Google Scholar
28. Holcomb HH, Cascella NG, Thaker GK, Medoff DR, Dannals RF, Tamminga CA: Functional sites of neuroleptic drug action in the human brain: PET/FDG studies with and without haloperidol. Am J Psychiatry 1996; 153:41–49Link, Google Scholar
29. Buchsbaum MS, Potkin SG, Marshall JF, Lottenberg S, Teng CY, Heh CW, Tafalla R, Reynolds C, Abel L, Plon L, Bunney WE Jr: Effects of clozapine and thiothixene on glucose metabolic rate in schizophrenia. Neuropsychopharmacology 1992; 6:155–164Medline, Google Scholar
30. Buchsbaum MS, Potkin SG, Siegel BV Jr, Lohr J, Katz M, Gott~schalk LA, Gulasekaram B, Marshall JF, Lottenberg S, Teng CY, Abel L, Plon L, Bunney WE Jr: Striatal metabolic rate and clinical response to neuroleptics in schizophrenia. Arch Gen Psychiatry 1992; 49:966–974Crossref, Medline, Google Scholar

